INTRODUCTION
Mazabraud syndrome (MS) is a disease defined by a combination of skeletal fibrous dysplasia (FD) and intramuscular myxoma (IM) [1]. The disease is extremely rare, with an incidence of only a few out of a million people. So far, only a hundred cases have been reported in the literature. FD is characterized by the destruction of bone tissue and replacement with a dysplastic proliferation of fibrous tissue and immature woven bone. The systemic manifestations of FD may present with endocrine dysfunction and cutaneous hyperpigmentation [2].
Soft tissue myxoma is a benign tumor of mesenchymal origin. In the case of MS, they usually occur when the patient is relatively older and in close vicinity to the bones most severely affected by FD. In addition, it is reported that MS is rarely associated with McCune-Albright syndrome (MAS). MAS is a rare syndrome that includes a combination of three characteristics: polyostotic FD, café-au-lait spots, and endocrine dysfunction. Twenty-six patients with MS associated with MAS have been reported, of which only 10 had the typical combination of some clinical features of MAS [3]. In this case, we report an extremely rare MS case that had some characteristic features of MAS.
CASE REPORT
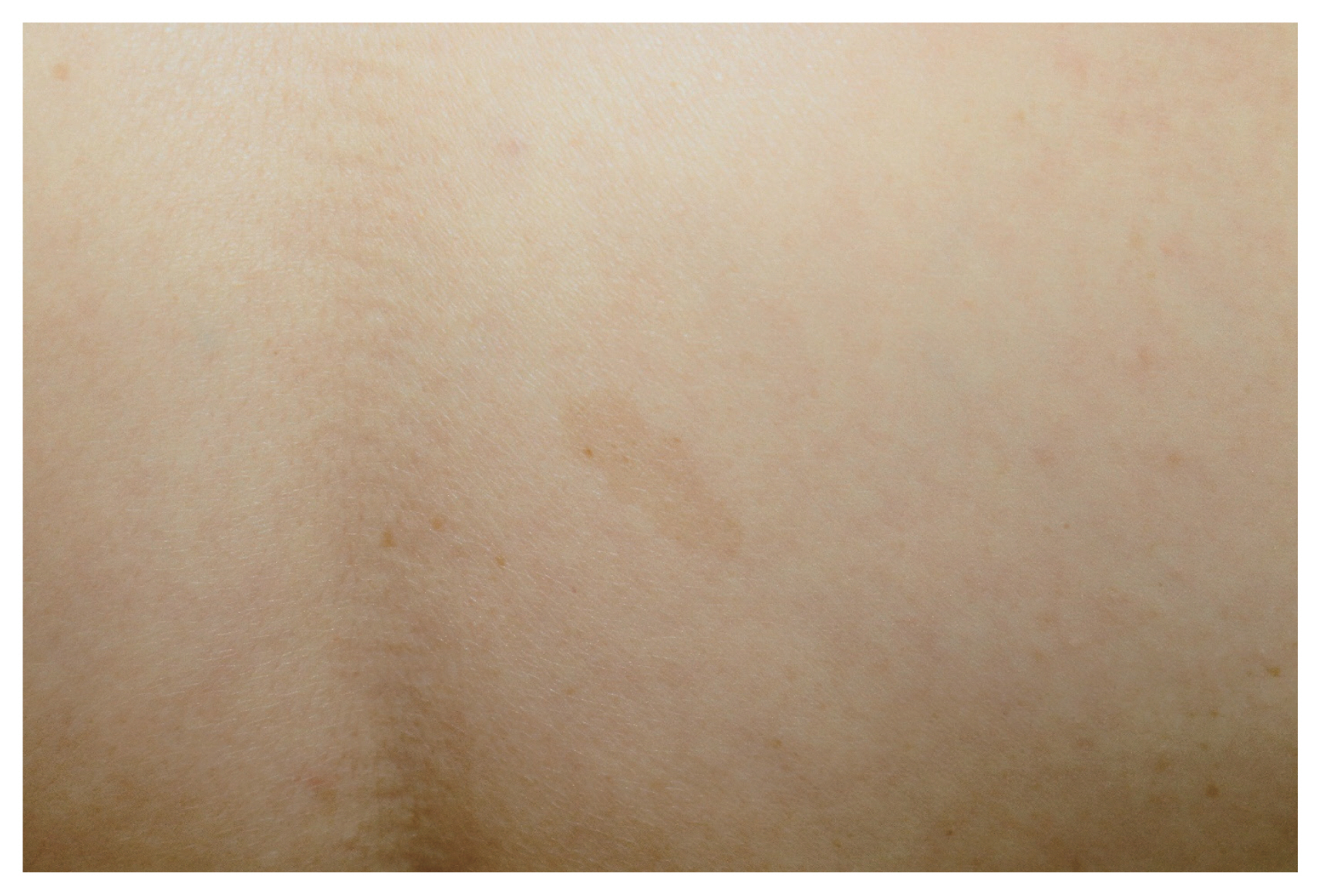
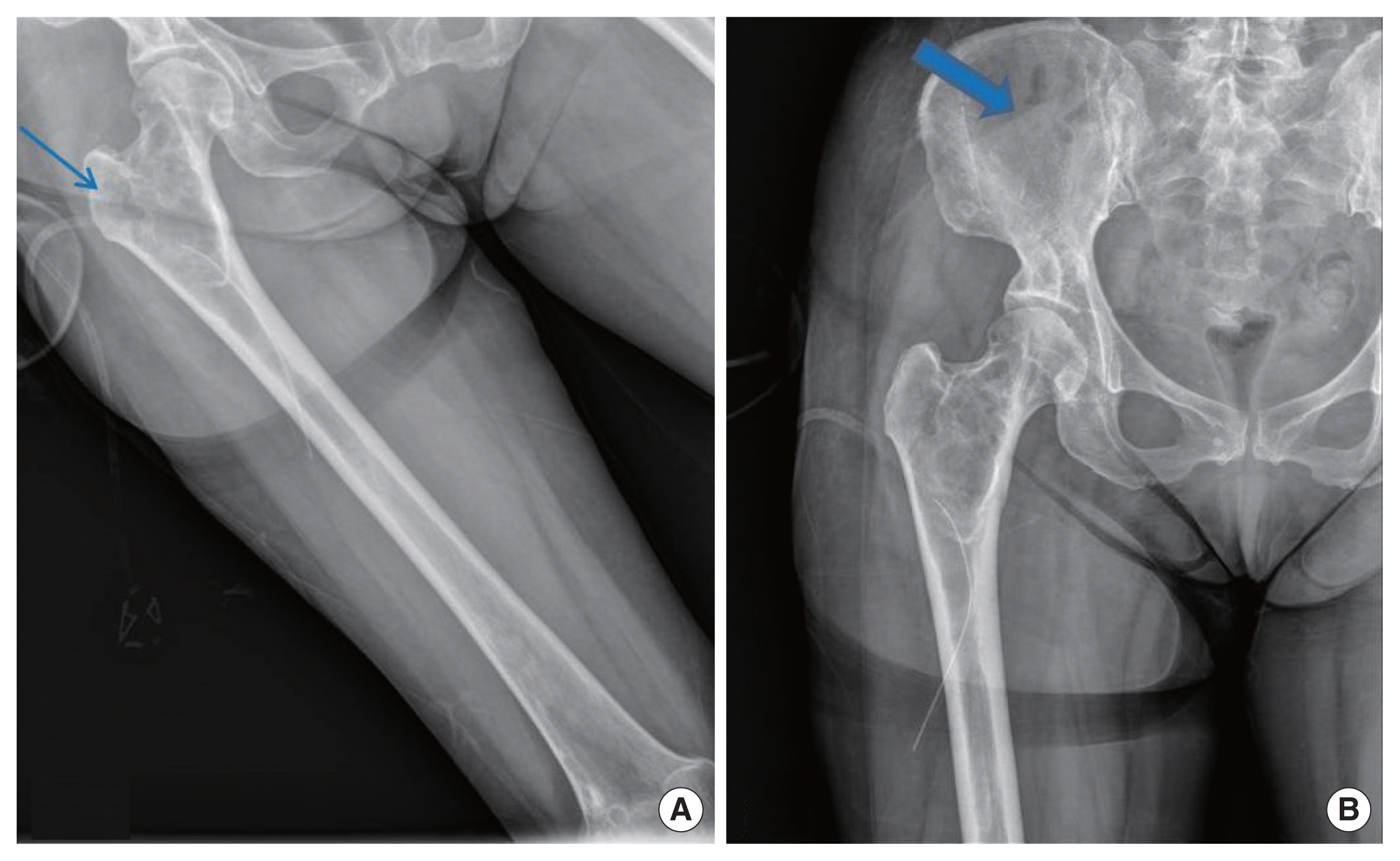
A 59-year-old female patient came to the outpatient department with a non-tender mass of the right thigh that grew from 3 years ago. The mass was about 5 cm in size, smooth and easily movable, round-shaped, and located on the anterolateral side of the thigh. There was no trauma history around the lesion. This patient had menstruation onset before age 13 and was taller in adolescence than her peers. Upon physical examination, multiple café-au-lait spots were observed on both the forearms and back (Fig. 1). On magnetic resonance imaging (MRI), the right vastus lateralis muscle area showed a mass with well-defined heterogeneous enhancement, accompanied by an inner cystic lesion (Fig. 2). Similar lesions were also observed in the femur neck and trochanteric area. This mass showed a slightly lower signal intensity than the surrounding muscle tissue on T1-weighted images and heterogeneously high signal intensity on T2-weighted images. After an injection of gadolinium, the mass was heterogeneously contrast-enhanced. Simple plain films revealed multiple lesions, which showed minimal endosteal scalloping cortical thinning, the loss of the normal trabecular pattern, and a ground-glass appearance in the right femur and ilium (Fig. 3). These radiographic findings were consistent with polyostotic FD with multiple bone invasions. Based on a suspicion of MS, an excisional biopsy of the mass was performed.
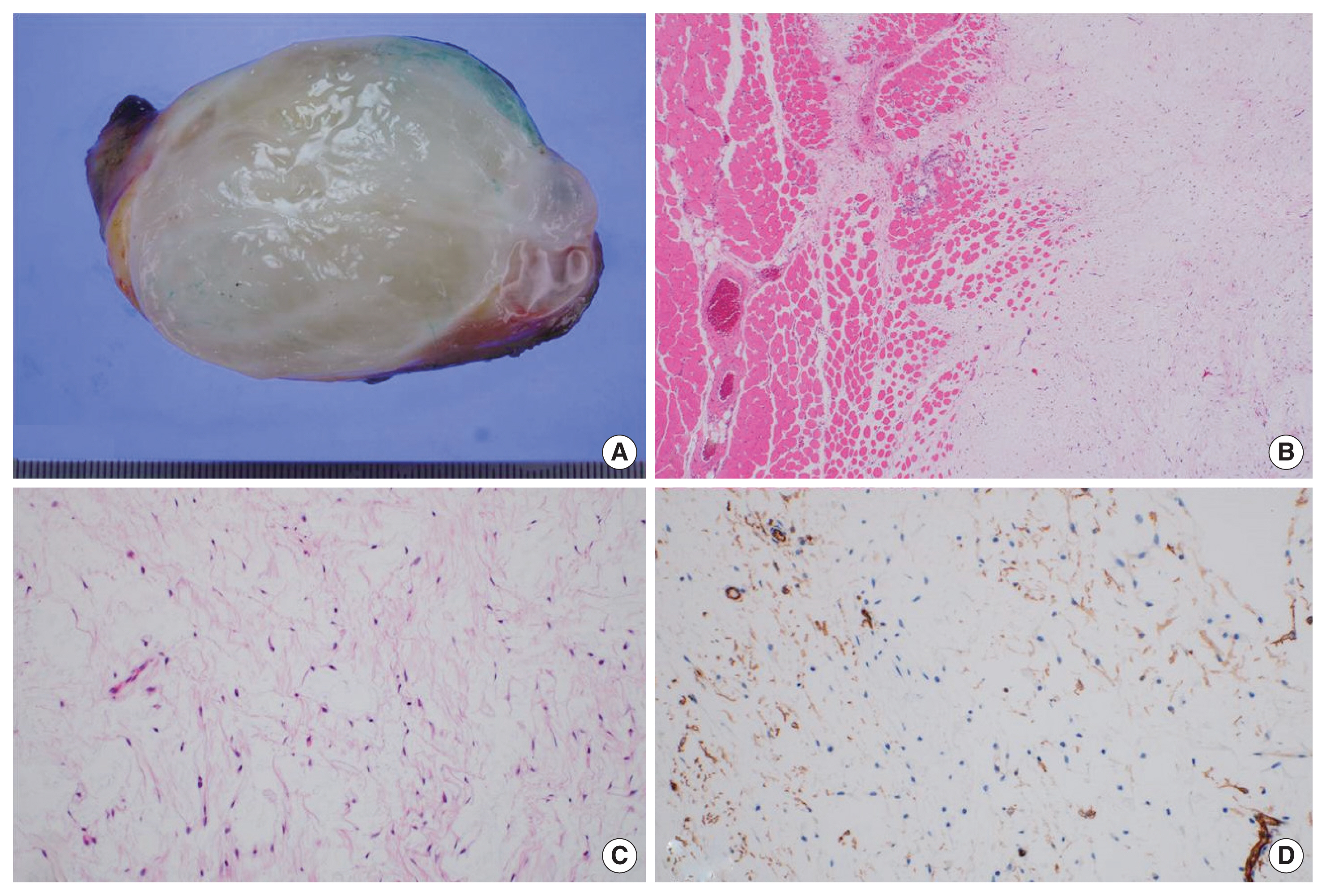
Three masses were found in the intraoperative field and were encapsulated and surrounded by skeletal muscle. The masses were located in the intramuscular layer beneath the vastus lateralis muscle, and all showed similar gross appearances. They were reddish, very soft masses, 6.0×4.5 cm, 4.0×3.0 cm, and 2.0×1.5 cm in size. Cross-sections of the tumor revealed relatively, well-demarcated gray-white gelatinous tumors (Fig. 4A). Histologically, the tumors were ill-marginated with entrapment of adjacent skeletal muscle fibers (Fig. 4B) and were hypocellular lesions with abundant myxoid stroma (Fig. 4C). There were scattered small blood vessels within the tumors. Immunohistochemical staining for CD34 and SMA was positive and staining for EMA, S100, and desmin was negative (Fig. 4D). The tumors were poorly vascularized, and neither hemorrhage nor necrosis was observed in the specimens. The patient was diagnosed with IM. No intra-lesional excision or cryosurgery of the right proximal femur tumor was performed because the patient had no pain during walking and strongly refused a bony biopsy. The patient was discharged on the sixteenth day after surgery without specific complications. such as gait disturbance or claudication.
This study is a retrospective case review. All medical records in the study involving human participants were reviewed in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards. Informed written consent was obtained from the patient for publication of this report and any accompanying images.
DISCUSSION
MS is a rare but well-characterized disease. A combination pattern of FD and IM was described by Mazabraud et al. [1] and first defined as MS in 1967. The symptoms are related to the two different diseases involved. However, there may be pathological fractures and pain, or the size of the painless mass may increase.
FD in itself is not a rare disorder; it is reported to account for 5%–7% of benign bone tumors [4]. Monostotic FD presentation (affecting only one bone) is more frequent than the polyostotic type (appearing on more than one bone). FD is thought to occur as a result of a developmental failure in the remodeling of primitive bone to mature lamellar bone and a failure of the bone to rearrange in response to mechanical stress. For this reason, pain, gross deformity, and pathologic fractures occur in FD. FD most often occurs in the order of the femur, tibia, and skull [5].
IM appears mainly as a single lesion and rarely shows accompanying clinical abnormalities. IM is hypovascular, never an epithelial component, and usually does not recur locally [4]. Several other benign lesions, such as mucinous neurofibroma and myxochondroma, and malignant mesenchymal lesions, such as mucinous liposarcoma should also be considered for differential diagnosis of IM [6].
The relationship between FD and IM has not been clarified. According to recent literature, a common histogenesis has been proposed for the two diseases. Wirth et al. [7] suggested a basic metabolic error in the bone growth area of the two diseases in the early stages of growth. Other authors have demonstrated that GNAS1 mutations exist in IM, which may also play a crucial role in the tumorigenesis of IM and FD. Mutations in the GNAS1 gene also appear to be another characteristic of FD in MAS. Cohen and Howell [8] hypothesized that somatic mutations in small cell masses likely result in FD in MAS. Mutations in larger cell masses can cause polyostotic FD [8].
In MS, the polyostotic form of FD is more common than the monostotic type. In general, the occurrence of FD is often preceded by IM, and IM of the soft-tissue lesions becomes apparent many years later, usually in the fifth or sixth decade of life. FD of the bones occurs mainly in the first 3 decades of life [5]. In 2019, Vescini et al. [9] reviewed all 106 MS patients reported since the first report. Interestingly, FD developed an average of 6.5 years earlier than IM lesions. And, café-au-lait spots and precocious puberty characteristics were reported in 10 patients [9]. The affected areas of the IM are mainly the large muscles of the thigh. IM may be certainly diagnosed based on the presence of a typical blend of spindle cells and stellate-shaped cells with small nuclei in an abundant extracellular myxoid stroma. In the case of FD, as a unique finding, spindle cells or fibrous tissues that can be seen only in myxoma other than osseous tissue, are observed.
A solitary myxoma can prevent the misdiagnosis of a malignant mesenchymal tumor containing myxoid tissue. For patients with multiple intramuscular tumors associated with bone lesions, it is important to recognize MS to avoid misdiagnosing them as malignant tumors and performing wide excision and radiotherapy. In addition, a proper histopathologic confirmation should also be made to exclude malignant tumors, keeping in mind the possibility of a sarcomatous transformation or a metastatic tumor in an elderly patient. The frequency of malignant changes in FD in MS is higher than that in FD without intramuscular myxoma [10].
In the case of MS patients, IM, which usually causes relatively fewer symptoms, is often diagnosed at the same time as the onset of FD or later because the first visit is usually due to the discomfort caused by FD. Usually, IM lesions appear years after the onset of FD, so a careful long-term follow-up is suggested for MS patients. Therefore, in the case of patients diagnosed with IM, it is important to make sure that FD is not visible in the osseous lesions, such as those in the thigh, if MS occurs as well.
There were some limitations to this case. First, the patient failed to undergo a bone biopsy, so the pathological characteristics of FD were not confirmed. In addition, evaluations of endocrine dysfunction which are characteristic of MAS, were not performed. Had the hormones been properly tested, it would have been possible to report a rarer case in which MAS and MS appeared simultaneously. Finally, it was also a limitation that genetic analysis of the GNAS1 gene in the myxoma or bone cells, which has been identified as the cause of MS, was not performed.
Because the association between FD and IM is rare, it is thought that each disease is often misdiagnosed and could not be properly diagnosed as MS. At our clinic, we initially focused only on mass removal and did not adequately examine the bone lesions. After the complete excision of the thigh mass, MAS was added to the presumptive diagnosis, and café-au-lait spots were also observed. MS is a rare but well-characterized disease, so considering it may reduce the risk of misdiagnosis. In addition, in the case of FD with IM, the risk of malignant changes in bone tumors increase. Therefore, patients with any of the characteristics of FD and IM should be examined for MS in impression.