INTRODUCTION
Allogeneic hematopoietic stem cell transplantation (HSCT) is frequently performed in patients with hematologic malignancies. A pre-transplant screening work-up of donors for allogeneic HSCT is essential in an effort to minimize risks to the recipient and to protect the donor. Recommended donor work-ups include a complete medical history and physical examination, blood workup (complete blood count, differential counts, complete panel, lactate dehydrogenase, prothrombin time/partial thromboplastin time, blood type, human leukocyte antigen (HLA) testing, and β-human chorionic gonadotropin), infectious disease testing (cytomegalovirus immunoglobulin G, Epstein-Barr virus, human immunodeficiency virus-1 and -2 antibody, human immunodeficiency virus antigen nucleic acid amplification test, human T cell leukemia virus-1 and -2 antibody, West Nile virus nucleic acid amplification test, hepatitis B surface antigen, hepatitis B core antibody, hepatitis C virus nucleic acid amplification test, rapid plasma reagin, and type and screen), urinalysis, chest X-ray, and/or electrocardiography. Despite routine screening, inadvertent transmission of various malignant and nonmalignant diseases from donor to recipient has been reported in several cases [1–3]. Kiss et al. [4] suggested that bone marrow aspirates, as part of routine donor assessment for allogeneic HSCT, reveal the presence of occult hematological malignancies in otherwise asymptomatic individuals. However, routine bone marrow and karyotype examination of donors is still not carried out because of cost-ineffectiveness and discomfort for the donor. We report the case of a patient with constitutional cytogenetic abnormality transmitted from an allogeneic stem cell donor.
CASE REPORT
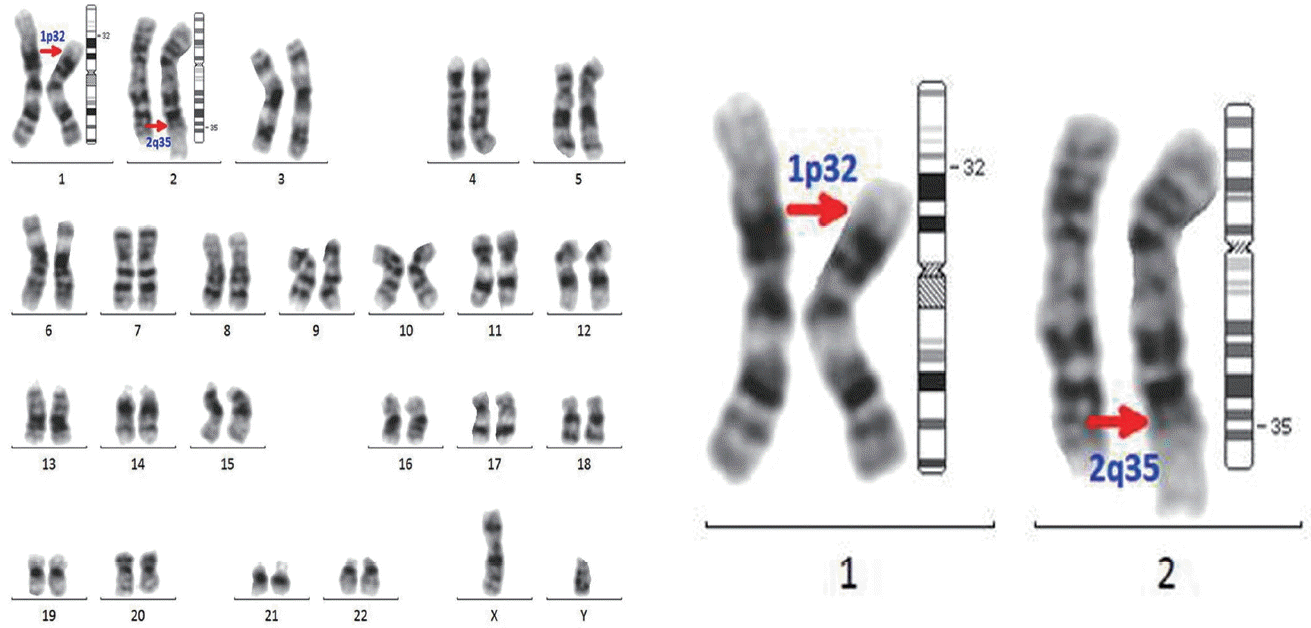
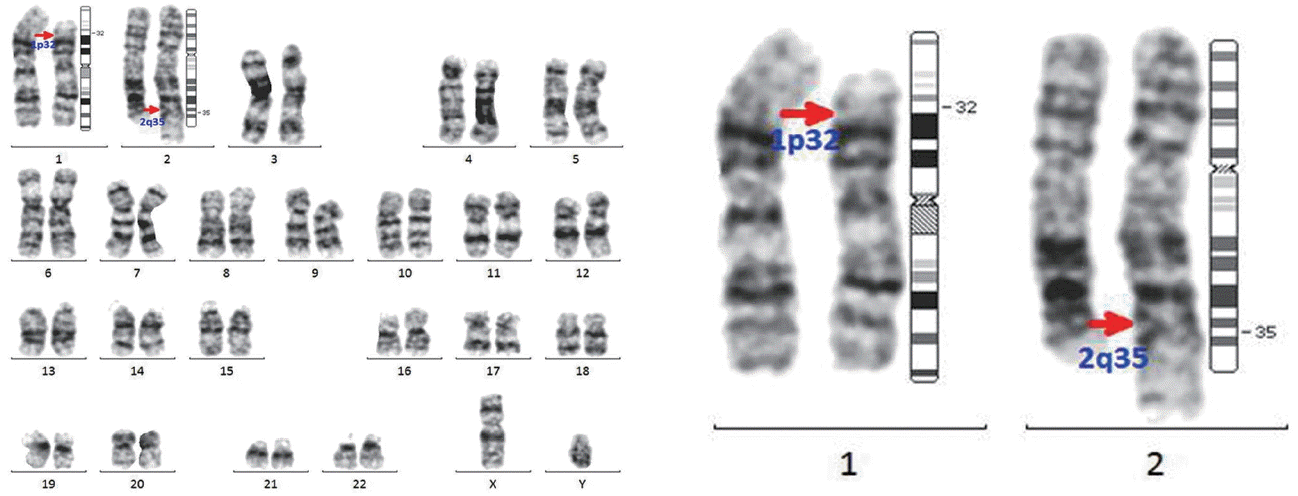
In December 2009, a 49-year-old man was diagnosed with acute myeloid leukemia (AML) with t(8;21)(q22;q22), AML-1/eight twenty one (AML1/ETO), and clonal evolutions (46,XY,t(8;21)(q22;q22){11}/45,idem,−Y{5}/47,idem,del(9)(q22),+21{4}/46,XY{1}). He underwent a course of induction chemotherapy with conventional idarubicin and cytarabine and achieved cytogenetic complete remission (CR) (46,XY[20]) (Fig. 1). He was treated with an additional cycle of consolidation chemotherapy consisting of high-dose cytarabine followed by allogeneic HSCT from a fully matched sibling donor, due to additional cytogenetic abnormalities, including deletion in 9q and trisomy 21, which are known adverse prognostic factors. The donor was a related 44-year-old male with no reported medical history. The patient underwent a conditioning treatment consisting of busulfan and fludarabine and received a peripheral blood stem cell transplant containing 1.6×106 CD34+ cells/kg. Prophylaxis for graft-versus-host disease (GVHD) consisted of tacrolimus and a short course of methotrexate. Engraftment was achieved on day 12, and a bone marrow examination on day 21 confirmed cytogenetic CR. A histological evaluation revealed no evidence of leukemia by morphology or flow cytometric analysis. A conventional cytogenetic study identified the presence of 46,XY,t(1;2)(p32;q35) in all 20 cells examined (Fig. 2). A chimerism analysis using multiplex polymerase chain reaction to amplify an informative short tandem repeat at the D21S11 locus was demonstrated in 98.6% of donor cells. Fluorescent in situ hybridization (FISH) analysis was performed, which did not show the AML1/ETO rearrangement in 500/500 cells examined. Given that the chimerism data showed 98% donor hematopoiesis, there was a high level of suspicion that the t(1;2) aberration was of donor cell origin rather than a previously unidentified, or newly acquired, clone of recipient cell origin. An aliquot of the donor graft was cryopreserved before infusion into the recipient. A phytohaemagglutinin-stimulated T lymphocyte culture was performed with this specimen, which revealed t(1;2) in all 30 cells examined (Fig. 3). On day 28 after transplant, the patient developed veno-occlusive disease, which was eventually brought under control with heparin therapy on day 42. A bone marrow biopsy and aspirate were repeated on day 105 after transplant with similar morphologic and engraftment findings. Cytogenetics on this sample detected 46,XY,t(1;2)(p32;q35) in all 20 cells examined. His course was complicated on day 187 by invasive pulmonary aspergillosis, which was controlled with voriconazole. During these complications, the patient was noted to be recurrently pancytopenic, requiring intermittent platelet and red blood cell transfusions. Ultimately, the patient regained normal hematopoiesis 6 months after allogeneic HSCT.
DISCUSSION
HSCT is increasingly used to treat a variety of malignant and nonmalignant hematological diseases. Advances in HLA typing, new immunosuppressive protocols, and better supportive care as well as the use of reduced conditioning protocols have improved outcomes of HSCT. However, there are still a number of problems to solve with regard to donor and recipient safety. In particular, an issue of vital concern is transmission of donor illness via stem cell transplantation. Niederwieser et al. [3] reported inadvertent transmission of donor AML in a bone marrow transplantation (BMT) for chronic myelocytic leukemia. Sevilla et al. [5] reported transient donor cell-derived myelodysplastic syndrome with monosomy 7 after unrelated cord blood transplantation. Berg et al. [2] described transmission of T cell lymphoma by allogeneic BMT. The malignant diseases identified in these recipients after transplant may have existed subclinically in the donors before transplant.
In addition to the transmission of malignant disease, donors with constitutional cytogenetic abnormalities have also been rarely reported. Manola et al. [6] reported two persons with mosaic Turner syndrome serving as stem cell donors. These donors had no overt symptoms; however, they had the potential to develop diabetes, osteoporosis, and immunologic disturbances. Frey et al. [7] reported a person with trisomy 8 who served as a stem cell donor. Given the rarity of trisomy 8 mosaicism (T8m), it is difficult to assess whether the presence of this constitutional abnormality is associated with an increased risk of hematologic malignancies. Several people with T8m have been reported to develop hematologic malignancies [8–10].
Our report is a case of inadvertent transmission of a donor’s constitutional t(1;2)(p32;q35) to a recipient. The donor with t(1;2)(p32;q35) received a clean bill of health, and there was no family history. His condition was considered an inclusion criterion for stem cell donation. In Korea, routine screening of adult donors does not include a bone marrow examination, so we did not know that he had constitutional cytogenetic abnormalities. The t(1;2)(p32;q35) aberration is a very rare cytogenetic abnormality, and its exact role is unclear. Several cases have been reported correlating chromosome 1 band p32 (1p32) aberrations and T lymphoblastic leukemia/lymphoma [11]. However, it is difficult to assess whether the presence of this constitutional abnormality is associated with an increased risk of malignancy.
Constitutional chromosomal abnormalities are inherited from a carrier parent or are present in all tissues after occurring de novo in gametes. Data from literature reviews have consistently shown a <1% incidence of all constitutional aberrations in hematologic malignancies, similar to that found in the general population [12,13]. These results suggest that persons with constitutional chromosomal abnormalities do not have an increased risk of hematologic malignancy. However, there are some reports of constitutional translocations with breakpoints similar to leukemia-associated specific breakpoints in hematologic malignancy [14]. Patients treated with allogeneic HSCT are not representative of the general population, as they are exposed to radiotherapy, chemotherapy, and even immune dysfunction (T cell depletion, HLA mismatch, GVHD, and immunosuppressive therapy); these are risk factors for acquiring genetic mutations. The hypothesis proposed by Knudson et al. [15] suggests that two mutational events are required for the development of cancer; the first step is a constitutional event and the second is an acquired genetic mutation. Inheriting constitutional t(1;2) can serve as a foundation for the development of secondary cancer after allogeneic HSCT.
These results suggest the importance of bone marrow and karyotype examination of donors. A cytogenetic study of all donors would allow the detection of abnormal clones and prevent transmission to other persons. In fact, few centers perform routine bone marrow aspirates as part of the donor work-up. However, to carry out these examinations systematically in all donors would be cost-ineffective, and it would be uncomfortable for the donors. Kiss et al. [4] asserted that more screening investigations, including bone marrow aspiration, might be reasonable to investigate for occult hematological malignancies prior to stem cell donation, particularly in older donors and donors with potential for familial malignancies. We suggest that more extensive donor screening, including bone marrow aspiration with karyotyping, may be reasonable if the recipient is young. Young patients have more time to be exposed to hazardous environments that can cause acquired genetic alterations. A less invasive method of examination with a simple procedure including detection of constitutional cytogenetic change by using oral mucosal cells could also be considered.
We report the first case of a person with constitutional t(1;2) serving as a stem cell donor. Although one case is insufficient to support the need for routine bone marrow biopsy prior to transplantation, the need for precise evaluation of the incidence, prognosis, and outcome of donor-transmitted chromosomal abnormalities is suggested. A nationwide registry of donor-transmitted abnormalities is necessary to allow for possible changes in donor screening.