INTRODUCTION
Common variable immunodeficiency (CVID) is a heterogeneous group of primary immune disorders presented with a defective immunoglobulin production with impaired immune response. Although there is no perfect consensus on the diagnostic criteria of CVID, it is usually defined when a patient has (1) markedly reduced serum immunoglobulin G (IgG) with low levels of immunoglobulin A (IgA), and/or immunoglobulin M (IgM), (2) poor or absent antibody response to infection or immunization, and (3) an absence of any other immunodeficiency state or secondary cause of hypogammoglobulinemia [1]. Although the number of B-cells in patients with CVID is usually normal, the B-cells have impaired immune function due to dysfunction of toll-like receptors on B-cells and the scarcity of isotype-switched memory B-cells [2]. In addition, T-cell abnormalities such as a low CD4/CD8 ratio were found in a subset of CVID patients and it is correlated with the severity of clinical manifestations [3].
Recurrent infection is the most common manifestation of patients with CVID and sinopulmonary infection is observed in the majority of patients [1,4]. Patients may also suffer from autoimmune disease, lymphocytic organ infiltration such as granuloma or splenomegaly, and malignancies [4]. Intravenous immunoglobulin (IVIg) replacement can dramatically reduce the incidence of death from bacterial infection [5]. However, IVIg replacement is not a curative approach and it is unclear that the replacement protects against granulomatous disease or cancers [5]. Here, we report a case of CVID followed by severe aplastic anemia (SAA) that successfully achieved resolution from propensity to infection as well as normal hematopoiesis by reduced intensity conditioning (RIC) allograft.
CASE REPORT
A twenty-year-old male patient presented with right chest wall pain for a week. He had developed recurrent sinopulmonary infection ever since he was three months old. When he was six years old, he suffered from severe pneumonia after vaccination for influenza and was hospitalized for a month. Thereafter he would often develop sinopulmonary infection at the change of seasons. The vital signs were: 100/60 mm Hg for blood pressure, 100/min for heart rate, 18/min for respiratory rate, and 37.8°C for body temperature. Complete blood cell counts (CBC) were hemoglobin 14.1 g/dL; white blood cell (WBC) 6,560/μL (36.9% neutrophils and 51.4% lymphocytes); and platelet (PLT) 138,000/μL. Chest X-ray showed subpleural bulle combined with reticulation in right lower lung field, suggestive of pneumonia. He was diagnosed with bacterial pneumonia combined with infected bullae and improved after several days of intravenous antibiotic therapy.
During the treatment, the attending pulmonologist paid attention to his past history and presumed immunodeficiency. During childhood he completed vaccinations for major infectious diseases including viral hepatitis B according to the guideline of the Korean Society of Pediatrics; however, he was negative for hepatitis B surface antibody. Serum immunoglobulins were measured and showed markedly decreased blood levels; IgG 8 mg/dL (reference range, 680 to 1,620 mg/dL), IgA 4 mg/dL (reference range, 84 to 438 mg/dL), and IgM 21 mg/dL (reference range, 57 to 288 mg/dL). A lymphocyte subpopulation study using flow cytometry was conducted: counts and percentages of B-cells (CD19+), T-cells (CD3+), and helper (CD4+/CD3+) or cytotoxic (CD8+/CD3+) T-cells were all within normal range. However, CD4/CD8 ratio was decreased (0.59; reference range, 1.5 to 3.0). Diagnosis of CVID was established and IVIg replacement therapy was initiated. 400 mg/kg of IVIg was administered intravenously every 4 weeks. Unfortunately, our patient was hospitalized with infectious episodes repeatedly despite regular IVIg administration.
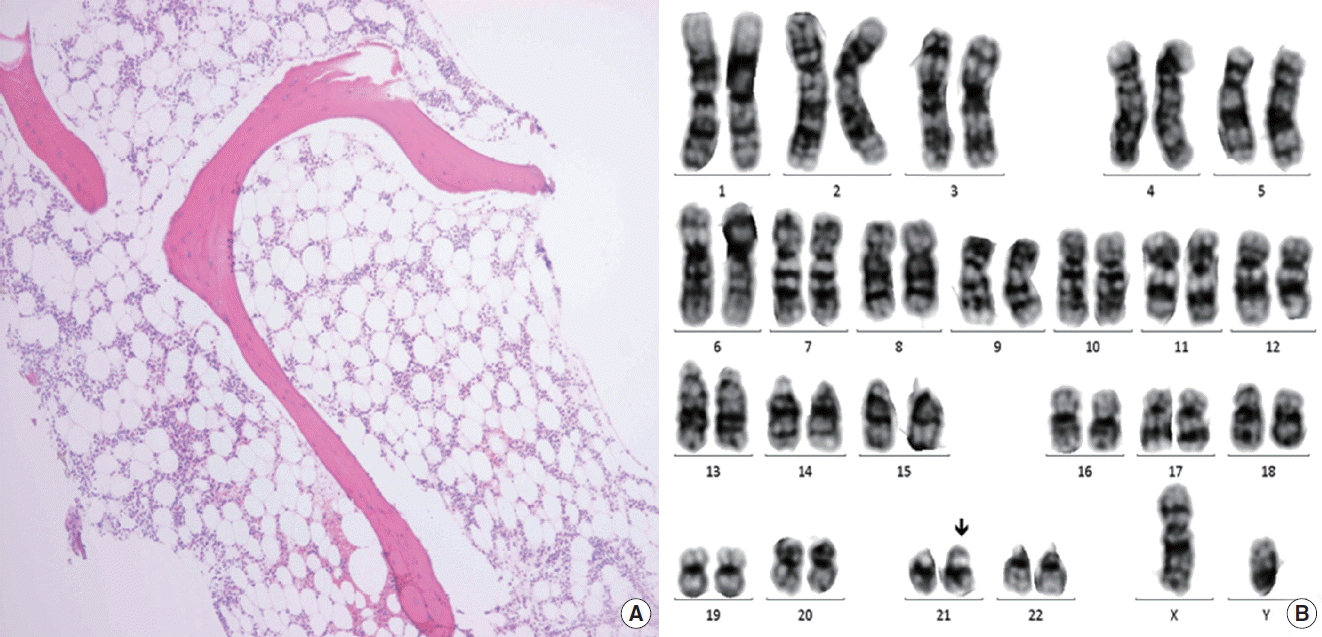
Eleven months after the diagnosis of CVID, he presented with petechiae on lower legs for three weeks. CBC showed pancytopenia: hemoglobin 8.8 g/dL; WBC 5,500/μL (19% neutrophils and 77% lymphocytes); and PLT 3,000/μL. Soon he became dependent on red cell and PLT transfusion with absolute neutrophil count <500/μL. Because pancytopenia was not resolved for a month, a bone marrow exam (BME) was performed and SAA was diagnosed according to the findings of decreased cellularity (<10%), no dysplastic changes or increase in blast cells, with morphologically normal residual hematopoietic cells along with normal karyotype (Fig. 1).
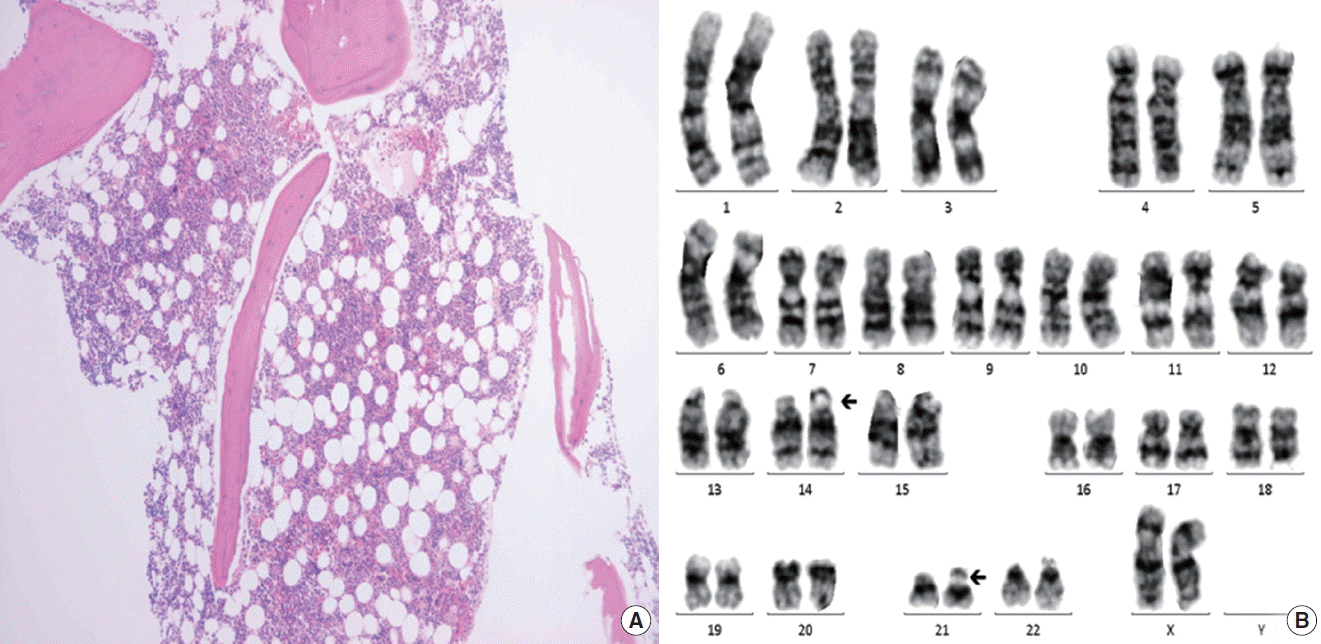
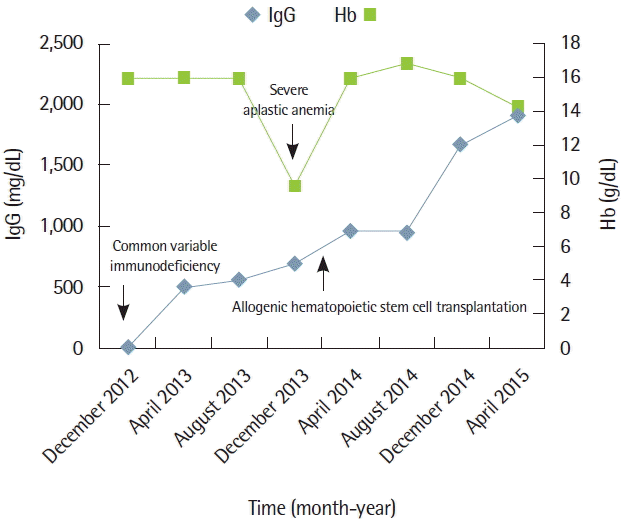
Allogenic stem cell transplantation (Allo-SCT) was considered for the treatment of SAA. He had a human leukocyte antigen-matched female sibling. We expected that vulnerability of infection caused by CVID might also be resolved after Allo-SCT by successful immune reconstitution after the procedure. Allo-SCT with RIC consisting of cyclophosphamide 60 mg/kg for 2 days and fludarabine 30 mg/m2 for 5 days in combination with antithymocyte globulin 2.5 mg/kg for 3 days was conducted. For the prevention of graft versus host disease (GVHD), methotrexate (15 mg/m2 on day +1 and 10 mg/m2 on days +3, +6, and +11) was administered in combination with intravenous tacrolimus (0.03 mg/kg/day from day −2 and doses were adjusted according to blood level and later changed to oral route). An engraftment was achieved successfully on day +12 and he was discharged from hospital on day +21. Follow-up BME (Fig. 2) and post-transplant short tandem repeat study performed on day +30 showed normal active trilineage hematopoiesis with complete donor chimerism. Grade 1 acute GVHD was developed on day +75 but it was confined to the skin and disappeared soon by topical steroids lotion and did not reemerge thereafter. Serum Ig G levels were normalized without IVIg administration after Allo-SCT. Immunosuppression was discontinued on day +120 and the patient has now maintained normal CBC and complete chimerism without any significant episode of respiratory infection for 17 months (Fig. 3).
DISCUSSION
Because Allo-SCT provides a new establishment of immune system derived from donor stem cells, it has been evaluated as a treatment option in primary immunodeficiencies [6]. The role of Allo-SCT in pediatric patients with severe combined immunodeficiency is well known and now the survival rate for Allo-SCT from matched sibling donor is probably over 90% [6]. Allo-SCT has also been applied to various congenital, early onset diseases of primary immunodeficiency such as Wiskott-Aldrich syndrome, congenital T-cell defects, phagocytic cell disorders, and hemophagocytic syndromes [6].
The majority of patients with CVID are diagnosed between ages of 20 and 45 [4]. This relatively late time of diagnosis resulted in a slightly different approach to treating CVID patients from those with early onset primary immunodeficiencies. IVIg replacement, the cornerstone of CVID treatment dramatically reduced the number of infectious events and antibiotic use. Busse et al. [5] reviewed the incidence of pneumonia among 50 patients with CVID. The annual incidence was 89% before the initiation of IVIg replacement. However, it dropped to 46% after the replacement over a mean period of 6.6±5.2 years [5]. Despite the improvement, IVIg replacement cannot perfectly prevent patients from infectious episodes and in many cases infection is still problematic even with IVIg therapy [4]. Furthermore, repetitive IVIg administration is a kind of maintenance therapy rather than a curative treatment. Therefore, a cessation of the replacement causes a decrease in IgG level and subsequent increased propensity to infection.
Early experiences of Allo-SCT in patients with CVID were derived from a subset of patient with CVID-related complications, especially non-Hodgkin lymphoma (NHL) or granulomatous lung disease. Rizzi et al. [7] reported outcomes of Allo-SCT from four patients with CVID (two patients with NHL and the other two patients with chronic lung disease). Three patients including two NHL patients survived with a durable complete remission of NHL or improved respiratory function and one of the three survivors was cured from CVID [7].
In our patients, development of SAA necessitated Allo-SCT. To our knowledge, there is only one previous report of any patient with both CVID and SAA: recently the Inborn Errors Working Party of the European Society of Blood and Marrow Transplantation (EBMT) reported multicenter experience in Allo-SCT for 25 patients with serious complications of CVID [8]. They reported that one of them had SAA and was indicative of Allo-SCT. The patient had a matched unrelated donor and received Allo-SCT with RIC using busulfan and melphalan [8]. However, detailed information including doses of conditioning regimen, GVHD prophylaxis, or follow-up duration after Allo-SCT was not provided in the report [8]. Although there was a report that some cases of CVID may be due to a mutation in the SBDS gene of Shwachman-Diamond syndrome [9], our patient had a feature of acquired SAA. However, we believe that it is natural to regard SAA as a kind of ‘immune disorders’ that can be observed in patients with CVID.
The multicenter analysis from EBMT reported that overall mortality was 52% and severe GVHD (32%) was a major affecting factor contributing to the relatively high mortality, possibly because for most patients only a single agent was used for GVHD prophylaxis [8]. High incidence of GVHD might be related to the increased serum tumor necrosis factor levels which are also observed in patients with CVID [10]. This report gives us insights into Allo-SCT for the treatment of CVID, including caution for GVHD. However, it was a retrospective analysis and the time of Allo-SCT ranges from 1993 to 2012 [8], suggesting that actual outcome might be better nowadays using improved GVHD prophylaxis and other supportive care.
In summary, Allo-SCT is a potentially curable option in patients with CVID complicated by SAA because both recovery of hematopoiesis and reconstitution of the immune system can be achieved by Allo-SCT if adequate efforts to minimize Allo-SCT-related complications accompany the treatment.