INTRODUCTION
Diabetic ketoacidosis (DKA) is a serious complication of adolescent diabetes mellitus (DM). When it comes down to diagnosis of type 2 DM in children, the rate of showing a presentation of DKA is relatively low, but it has often been reported in recent case studies [1]. On the other hand, diabetes with extreme hyperglycemia and hyperosmolality without ketosis is comprehended as hyperglycemic hyperosmolar state (HHS). HHS has a high mortality and morbidity rate in elderly patients with type 2 DM, however, has not been significantly recognized in young patients [2]. Although HHS is a distinct clinical entity from DKA, the two conditions may occur simultaneously, upbringing some treatment dilemma for practitioners. The following condition is currently diagnosed with the mixed HHS and DKA.
We report the rare case of an adolescent patient with type 2 DM who presented with mixed HHS and DKA.
CASE REPORT
A 15-year-old, previously healthy boy presented to the emergency room with a 4-day history of progressively increasing headache and dizziness. Two months before admission, he experienced weight loss of 15 kg, polyuria, and polydipsia. Further history revealed that he had often been consuming soda containing sugar due to thirst for approximately 1 year before presentation. His mother and grandmother had type 2 DM.
On admission, He had a blood pressure of 145/83 mm Hg and a pulse rate of 124 per minute. His height was 169 cm and his weight was 85 kg, yielding a body mass index of 29.4 kg/m2 and a waist circumference of 100 cm.
On physical examination, he was drowsy (glasgow coma scale [GCS] score, 12) and his breath smelled strongly of acetone. He had typical buffalo-humps around the neck and a moon-face. Acanthosis nigricans was present on the lateral side of his neck and in both axilla. Skin turgor was decreased and purple striae were present over the abdomen (Fig. 1).
Initial laboratory tests yielded a serum blood glucose level of >1,200 mg/dL and the following venous blood gas values: pH, 7.19 (reference range, 7.35 to 7.45); HCO3−, 6.9 mmol/L; and base deficit, −21.3 mmol/L. Electrolyte values were as follows: sodium, 138 mmol/L (reference range, 136 to 145 mmol/L) (corrected serum sodium, 155.6 mmol/L); potassium, 5.9 mmol/L (reference range, 3.5 to 5.1 mmol/L); and chloride, 96 mmol/L (reference range, 96 to 110 mmol/L). Calculated anion gap increased to 35.1 (reference range, 8 to 16). Blood urea nitrogen (BUN) was 44.5 mg/dL (reference range, 8.0 to 20 mg/dL); creatinine, 1.8 mg/dL (reference range, 0.5 to 1.1 mg/dL); osmolarity, 391 mosm/kg (reference range, 275 to 295 mosm/kg) (effective osmolarity, 343 mosm/kg); hemoglobin A1c, 11.4% (reference range <5.7%); amylase, 185 U/L (reference range, 8 to 110 U/L); total cholesterol, 305 mg/dL (reference range, 113 to 197 mg/dL); triglyceride, 467 mg/dL (reference range, 40 to 163 mg/dL); low-density lipoprotein (LDL) cholesterol, 199 mg/dL (reference range, 62 to 130 mg/dL); high-density lipoprotein cholesterol, 35 mg/dL (reference range, 35 to 65 mg/dL); C-peptide, 2.48 ng/mL (reference range, 0.48 to 3.3 ng/mL); free thyroxine, 1.4 ng/dL (reference range, 0.8 to 2.5 ng/dL); thyroid-stimulating hormone, 4.48 μIU/mL (reference range, 0.5 to 6.3 μIU/mL); C-reactive protein, negative; and anti-insulin antibodies, islet cell antibodies, and glutamic acid decarboxylase antibodies, all negative. Urine analysis revealed glucose 4+; ketone 3+; and protein 1+.
The patient was admitted to the intensive care unit and received fluid resuscitation with intravenous normal saline during the first hour followed by continuous insulin infusion starting at a rate of 0.1 IU/kg/hr. Blood pressure was 154/85 mm Hg and we started treatment with anti hypertensive agents using calcium channel blockers. The urine output remained greater than 1 mL/kg/hr and serum glucose slowly decreased to 751 mg/dL at approximately 90 mg/dL per hour for the first 5 hours.
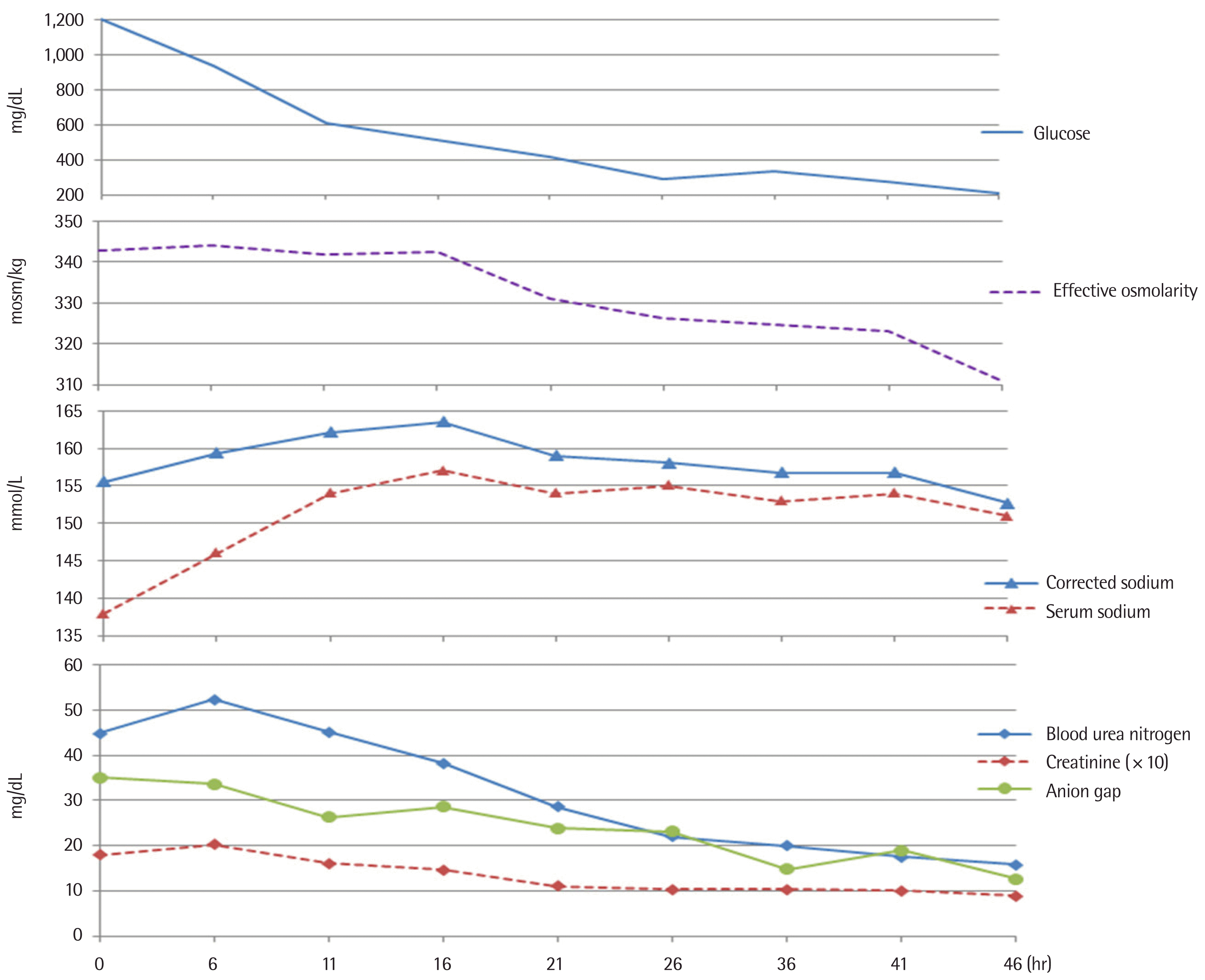
After 26 hours of management, he was alert (GCS 14), his dizziness and headache had subsided, and the purple striae on his abdomen became faint. His serum glucose gradually decreased to 292 mg/dL and his anion gap declined to 23.2. His serum BUN and creatinine had gradually increased up to 52.3 mg/dL and 2.04 mg/dL, respectively, in the first 4 hours but thereafter steadily declined to 22.1 mg/dL and 1.05 mg/dL, respectively, after 26 hours. Fractional excretion of sodium was calculated as 0.2% and urine output was maintained at 1.8 mL/kg/hr. While the serum glucose level was being corrected, the measured serum sodium increased to 157 mmol/L. Because the measured serum sodium was affected by hyperglycemia, increased measured serum sodium did not represent aggravation of existing hypertonic state. We used the calculated corrected sodium and its level of the patient was risen to 164 mmol/L, which then decreased steadily to 153 mmol/L at a slow rate thereafter. There was a small drop in effective osmolarity. Because follow-up serum levels of amylase and lipase for his initial elevated serum amylase increased to 522 U/L and 945 U/L, respectively, an abdominal computed tomography (CT) scan was performed to rule out pancreatitis.
On the third day, after he had recovered to an alert mental status (GCS 15) and his anion gap was normalized to 15.8, we permitted oral intake and transitioned from intravenous to subcutaneous insulin. Laboratory findings were improved as follows: serum glucose, 209 mg/dL; effective osmolarity, 311 mosm/kg; corrected sodium, 146 mmol/L, BUN, 15.9 mg/dL; creatinine, 0.9 mg/dL; amylase, 131 U/L; and lipase, 181 U/L (reference range, 0 to 60 U/L) (Fig. 2). Brain magnetic resonance imaging revealed no pathologic findings such as thrombosis or infarction. He resumed a regular diet and started to control his blood sugar level by injections of insulin glargine at night and insulin as part at every meal.
He was finally diagnosed with extreme obesity complicated with type 2 DM resulting in a mixed HHS and DKA. For evaluating complications of type 2 DM, nerve conduction tests, 24-hour urine collection, and an ophthalmology consultation were performed and mild proteinuria (7.93 mg/m2/hr) was identified.
For his cushingoid features, an evaluation for Cushing syndrome was performed. His adrenocorticotropic hormone (ACTH) level was 4.3 pg/mL (reference range, 10 to 60 pg/mL), cortisol was 55 μg/dL (reference range, 3 to 21 μg/dL), and his 24-hour urinary free cortisol excretion level was 209.6 μg/day (reference range, <84 μg/day). We planned a follow-up study and he was discharged after completing diabetes education.
One month after discharge, his laboratory tests were improved as follows: serum glucose was 154 mg/dL; total cholesterol, 166 mg/dL; triglyceride, 141 mg/dL; LDL cholesterol, 103 mg/dL; 24-hour urine protein, 1.65 mg/m2/hr (reference range, <4 mg/m2/hr); ACTH, 54.2 pg/mL; cortisol, 13.6 μg/dL; and 24-hour urinary free cortisol, 21.8 μg/day. His hemoglobin A1c improved 6.0% after a 3-month interval.
DISCUSSION
DKA and HHS are the two most serious and independent acute hyperglycemic crises of diabetes. Although HHS is a distinct clinical entity from DKA, the two conditions may occur simultaneously, resulting in some treatment dilemmas for practitioners.
Our patient had a presentation of mixed HHS and DKA as an initial manifestation. He had plasma glucose >600 mg/dL, osmolarity >320 mosm/kg, and hypernatremia, that met the criteria for HHS, and arterial pH <7.3, bicarbonate <15 mmol/L, and positive urine ketone indicating DKA (Table 1). He had characteristic features of type 2 DM, including a family history of type 2 DM, acanthosis nigricans, extreme obesity, and an absence of autoantibodies [2].
When diagnosing type 2 DM in children, the rate of presentation with DKA is relatively low, but it can range from 6% up to 33% in some case studies [1]. A few cases of mixed HHS and DKA have been reported, and most of these cases have been misdiagnosed as HHS. Morales and Rosenbloom [3] reported on seven children, all of whom were initially considered to have type 1 DM and DKA, but all died of HHS with type 2 DM. In another report of four adolescents, all had mixed HHS and DKA [2]. In 120 children with type 2 diabetes, five children presented with mixed HHS and DKA [4].
In DKA, fluid therapy is intended to correct acidosis and dehydration and the rate of fluid administration should not exceed 1.5–2 times the usual daily maintenance requirement. It is known that more aggressive fluid therapy is required for elderly adults with HHS. But for children with HHS, careful fluid therapy is required because rapid changes in serum osmolality may cause cerebral edema similarly to DKA [5].
In children with mixed HHS and DKA, treatment must take into account the potential complications such as electrolyte imbalance, cerebral edema and ischemic injury. The patient’s circulatory status and fluid balance should be reassessed frequently because patients with mixed HHS and DKA have a higher risk of cerebral edema than those with HHS [5]. Our patient received careful fluid therapy at lower than twice the maintenance rate after initial fluid resuscitation. We kept our patient in a hypernatremic state for a few hours initially. This is because a large drop in effective osmolality and a failure to increase measured serum sodium levels during treatment are associated with cerebral edema [5,6].
As the clinical features of dehydration had been improved with fluid therapy, acute renal failure (ARF), hyperamylasemia, and hyperlipasemia were thought to be due to an ischemic injury from severe dehydration. The etiology is thought to be multifactorial, mostly attributable to hypovolemia and hypo-perfusion [7]. There was no clinical evidence of renal parenchymal disease or other etiology of ARF such as infection or medication in our case.
Similarly, it was demonstrated that nonspecific elevations of amylase or lipase can be frequently observed in 16% to 25% of patients with DKA but these patients had no clinical or CT evidence of acute pancreatitis [8]. Rapid recovery of amylase and lipase to the baseline after treatment is observed in most patients. Because the pancreas is one of the most susceptible organs to ischemic injury, the elevation in pancreatic enzymes is due to ischemic injury caused by poor splanchnic perfusion during HHS.
Our patient had free access to high-carbohydrate-containing fluids before presentation and showed a hyperosmolar state as the initial presentation. Carbonated drinks contain a large amount of sugar, which accelerates the hyperglycemic state, and high sodium levels, which strongly exacerbates hypernatremia. This may have affected the severity of presentation. A previous report of five children who presented with hyperosmolarity also had a history of large-amount intake of hyperosmolar fluids [9]. Thus, cautious history taking has provided some assistance in narrowing the differential diagnosis.
Although it is very rare in childhood, Cushing syndrome should be considered in pediatric patients with obesity, diabetes, and purple striae. Our patient had typical cushingoid features and an evaluation was done. Serum and 24-hour urine cortisol level were high during DKA, but were normalized after 1 month of follow-up. In DKA or HHS, there is a reduction in the overall effective action of circulating insulin combined with a concomitant elevation of counter-regulatory hormones, such as cortisol. This is considered to be a pseudo-Cushing state [10].
DKA and HHS may coexist, making the treatment difficult. With increasing rates of childhood obesity and pediatric type 2 DM, cases of mixed HHS and DKA are expected to occur more frequently than before. Physicians need to have a high index of suspicion. Children with the mixed type should be managed with lower amounts of fluid compared to adults, and we highlight the fact that fluid therapy intended to gradually correct the osmolarity and sodium is important to avoid cerebral edema.