Familial Dysalbuminemic Hyperthyroxinemia로 진단된 19세 남자 환자 증례보고
Familial Dysalbuminemic Hyperthyroxinemia by Albumin Gene Variant (R242H) in a 19-Year-Old Male: A Case Report
Article information
Trans Abstract
Familial dysalbuminemic hyperthyroxinemia (FDH) is the most common inherited cause of high serum total thyroxine (T4) levels in clinically euthyroid individuals. Mutant albumin coding gene (ALB) interferes free T4 assays and leads to discordant thyroid function tests. We describe a 19-year-old male harboring a heterozygous c.725G > A (p.Arg242His) variant using Sanger sequencing for his ALB, which is the second FDH case reported in South Korea. Appropriate genetic testing for subjects suspicious of FDH would prevent unnecessary repeat tests, although the prevalence of FDH is very low in the Asian population.
서 론
Familial dysalbuminemic hyperthyroxinemia (FDH)는 albumin coding gene (ALB)의 변이로 발생하며 inherited euthyroid hyperthyroxinemia를 유발하는 가장 흔한 원인으로 알려져 있다. 유병률은 코카시아인에서 1만 명 중 1명 정도 진단되며, 프랑스인에서 0.08%, 덴마크인에서 0.01%, 아시아인에서 매우 드문 것으로 알려져 있다[1]. 우리나라에서는 본 저자가 ALB codon 242 c.725G > A (R242H) 변이를 가진 38세 여성을 국내 증례로는 처음 보고한 바 있다[2].
ALB 변이는 R242H가 가장 많이 보고되었고, 이외에도 R218P (c.725G > C), R218S (c.724C >A), R222I (c.737G > T)가 보고된 바 있다[1,3]. 이 변이들은 T4에 대한 결합 친화력(binding affinity)이 증가하여 혈청에서 주로 T4가 증가한다. 반면, L66P (c.269T > C)와 같이 T4가 아닌, T3의 결합 친화력이 증가하는 변이도 보고된 바 있다[1,3].
본 증례에서는 국내에서 보고하는 두 번째 FDH 증례로, 첫 번째 증례와 같은 R242H 변이가 발견되었다.
증 례
19세 남자가 다한증을 주소로 순천향대학교 부속 부천병원 내분비대사내과 외래에 방문하였다. 갑상선기능검사에서 혈청 free T4 3.78 ng/dL(참고범위, 0.8–2.0 ng/dL), T3 1.39 ng/mL(참고범위, 0.6–1.81 ng/mL), thyroid-stimulating hormone (TSH) 2.61 μIU/mL(참고범위, 0.39–5.44 μIU/mL)로, 혈청 free T4만 정상 상한치의 1.9배 상승한 소견을 보였다. 본원 갑상선기능검사는 Elecsys T3 kit, Elecsys FT4 kit, Elecsys TSH (Roche Diagnostics, Indianapolis, IN, USA)로 진행되었다. 다한증 외에는 특이증상 및 소견이 없었고, 뇌하수체 magnetic resonance imaging에서도 뇌하수체 종양 등 특이소견은 없었다. 환자가 갑상선질환 관련 유전자검사를 원하지 않아 추가 검사를 더 이상 진행하지 못 하였다.
2년 뒤 상기 환자는 외부 병원에서 무릎 수술을 받기 위해 검사하던 중 갑상선기능 이상소견으로 다시 본원으로 전원되었다. 외부 병원에서 메티마졸(methimazole) 30 mg을 처방받아 5주간 복용하였으나 갑상선기능에는 변화가 없었다. 내원 시 갑상선기능검사는 혈청 free T4 3.51 ng/dL, T3 1.12 ng/mL, TSH 2.37 μIU/mL로 여전히 혈청 free T4만 1.8배 상승한 소견이고 혈청 T3와 TSH는 정상 범위였다. 추가로 시행한 검사에서 혈청 T4 22.6 μg/dL(참고범위, 5.4–10.6 μg/dL), free T3 4.04 pg/mL(참고범위, 2.0–4.4 pg/mL)로 혈청 T4만 정상 상한치의 2.1배 상승하였고, 갑상선 자가항체는 음성이었다. 먼저 thyroid hormone receptor β (THRB) 변이에 대한 Sanger sequencing을 시행하여 음성(negative)의 결과를 얻었다. 이후 FDH를 확진하기 위해 BigDye Terminator Cycle Sequencing Ready Reaction kit (Applied Biosystems, Waltham, MA, USA)를 이용하여 ALB Sanger sequencing을 시행하였다. 말초 혈액 백혈구로부터 DNA를 분리하였고 ALB chromosome 4q13.3에 대한 직접 염기서열분석 결과, 725번째 염기인 G가 A로 치환되는 c.725G >A (p.Arg242His) 변이가 발견되어 FDH로 확진하였다(Fig. 1).
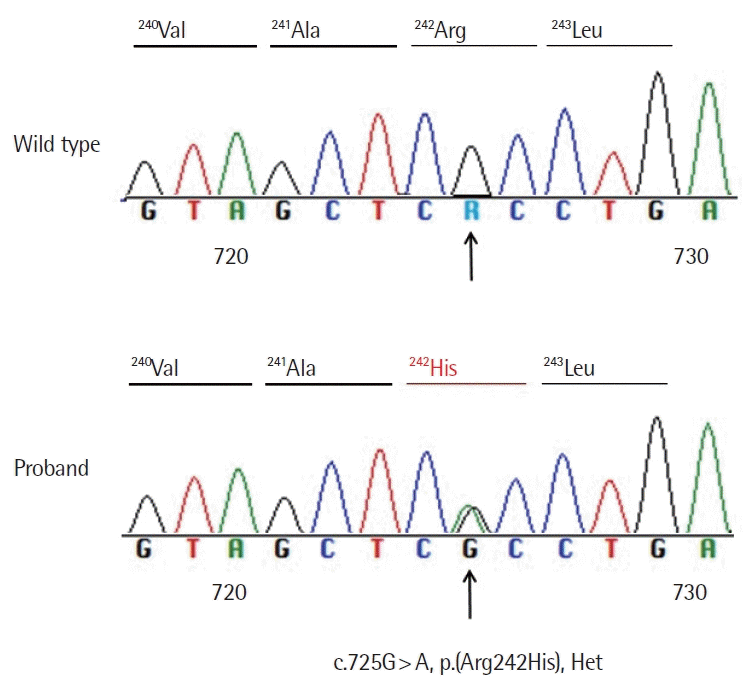
Sanger sequencing results for albumin coding gene.
환자는 임상적으로 정상 갑상선기능 상태였고, 갑상선기능 이상에 대한 추가 치료는 없었다. ALB 변이가 상염색체 우성 유전방식으로 유전되고 환자의 누나도 갑상선기능 이상으로 약제를 복용한 적이 있다고 하여 누나와 부모님이 본원에 방문하여 검사 받기를 권유했으나 내원하지 않았다. 본 증례를 보고하기 위해 환자에게 의무기록을 포함한 의학정보의 연구자료 이용 동의를 받았다.
고 찰
Syndrome of inappropriate secretion of TSH 소견이 있을 때 감별해야 하는 대표적인 질환이 syndrome of resistance to thyroid hormone (RTH)과 FDH이다. RTH는 여러 말초 조직에 분포하는 갑상선호르몬 수용체 α 또는 β의 유전적 변이로 갑상선호르몬 작용이 감소하는 질환으로 1967년 Refetoff 등[4]에 의해 처음 보고되었다. RTH 유병률은 4만 명 중 1명 정도로, 80% 이상에서 THRB 변이가 원인으로 알려져 있고 현재까지 약 3,000개의 증례가 보고되었다. RTH의 임상양상은 특이적이지 않고 다양한데, 갑상선종, 과잉행동, 빈맥, 발달 지연 등의 증상이 나타날 수 있다[5]. 97명의 THRB 변이를 가진 환자와 17명의 ALB 변이를 가진 환자의 임상 양상을 비교한 연구에서는 FDH 환자들에서 RTH에 비해 무증상의 빈도가 훨씬 높았다(59% vs. 14%, P = 0.0002). 반면, FDH 환자의 29%, RTH 환자의 67%이 갑상선기능항진증 증상을 호소하여, RTH 환자의 임상증상 발현빈도가 의미 있게 높았다(P = 0.007) [5]. 본 증례에서도 환자는 다한증 이외에 갑상선기능항진증을 시사하는 증상 및 갑상선종 등 특이소견이 없었고, THRB 변이의 Sanger sequencing을 통해 RTH를 배제하였다.
FDH 환자들은 시상하부-뇌하수체-갑상선 축으로 구성되는 갑상선호르몬 조절 기전에 잘 맞지 않는 갑상선기능 이상을 보이기 때문에 여러 병원에서 반복적인 검사를 하는 경우가 있다. FDH는 갑상선호르몬 운반을 담당하는 단백질 중 하나인 알부민의 유전적 변이로 발생하기 때문에 혈액검사에서 경도의 갑상선기능 이상을 보이지만 임상적으로는 정상 갑상선기능 상태이며, 별다른 치료가 필요하지 않은 경우가 많다. FDH 중 가장 많이 보고된 R242H 변이는 경한 형태(mild form)의 FDH로, T4에 대한 결합력이 10–15배 정도 증가하여 혈청 T4가 2배 정도 증가하는 것으로 알려져 있고[6,7], 본 증례에서도 약 2.1배 상승한 소견을 보였다.
FDH 환자의 혈청 free T4 수치는 갑상선기능검사 방식에 따라 다양하게 보고되고 있다. 2-step assay의 경우, T4 analogue capture 후 wash step를 통해 T4 analogue가 혈청 ALBumin과 결합하지 못하므로 이론적으로는 FDH 환자의 혈청 free T4가 정상범위에 머무르게 된다. 하지만 동일한 FDH 환자의 혈액을 이용해 다양한 1-step 또는 2-step assay로 혈청 free T4를 측정한 결과를 보면, 대체적으로 2-step assay는 참고범위를 약간 상회하는 결과를 보이고, 1-step assay는 참고범위보다 훨씬 높은 혈청 free T4 값을 보여 검사법에 따라 free T4 변동 폭이 크다는 것을 알 수 있다[8]. 임상에서 자주 사용하는 Immulite 2500 and ADVIA Centaur (Siemens, Forchheim, Germany), Elecsys E170 (Roche, Rotkreuz, Switzerland), Access (Beckman Coulter, Brea, CA, USA), AIA-1800 (Tosoh Bioscience, Tokyo, Japan)는 혈청 free T4가 참고범위를 훨씬 상회하는 결과를 보였으며, 이 중 Beckman Coulter를 제외하고는 모두 1-step assay이다. 2017년 보고한 국내 첫 FDH 증례에서도 Siemens, Beckman Coulter, Roche의 세 가지 kit로 혈청 free T4를 측정하였을 때 상당한 차이가 있었고, 그중 Roche로 측정한 값이 가장 높았다[2]. 본 증례에서도 Roche의 kit를 이용해 측정하였다.
FDH는 유전질환으로 유병률이 낮아 국내 보고로는 이번 증례가 두 번째이지만 실제로는 더 많은 증례가 있을 것으로 추정된다. TSH 반응이 적절하지 않은 갑상선기능 이상을 보일 때 FDH가 의심된다면 ALB 변이를 확인하는 확진검사를 통해 불필요한 반복적인 검사를 피할 수 있다.