쿠싱증후군으로 발현한 췌장내분비샘암종 1예
A Case of ACTH-producing Pancreatic Neuroendocrine Tumor Presenting with Cushing Syndrome
Article information
Trans Abstract
Pancreatic neuroendocrine tumors are rare neoplasm arising from pancreatic islet cells. Occasionally they are functioning tumors secreting a variety of hormones such as insulin, gastrin, glucagon, and vasoactive intestinal peptides. Adrenocorticotropic hormone (ACTH) secreting pancreatic neuroendocrine tumors (ACTHomas) are very rare and there have been about 110 case reports worldwide. Due to excessive ectopic ACTH production and resulting hypercortisolemia, patients with ACTHoma usually present with Cushing syndrome. ACTHomas have a poor prognosis with severe and rapidly progressive clinical courses. They often present with unresectable liver metastases and there remains little consensus on its standard treatment. We report a 55-year-old female with ACTH-producing pancreatic neuroendocrine tumor, who was treated with surgical resection of pancreatic tail, spleen, and a portion of stomach. Sixteen months later, abdomen computed tomography scan showed multiple liver metastases, which were treated with transarterial chemoembolization.
서 론
췌장의 신경내분비종양은 약 인구 십만 명당 1명 발병하며 전체 췌장암의 1-2%를 차지하는 드문 종양으로[1,2] 50대에서 70대에 가장 많이 발생하는 것으로 알려져 있다. 최근에는 복부 전산화단층찰영이나 내시경검사의 증가로 그 빈도가 늘어나고 있는 추세이다. 분화도에 따라 분화도가 좋은 신경내분비종양과 분화도가 나쁜 신경내분비종양으로 나뉜다. 호르몬 분비 여부에 따라 비기능성과 기능성으로 분류되며 약 70-80% 경우 비기능성으로 알려져 있다. 분비하는 호르몬의 종류에 따라서 인슐린종, 가스트린종, 글루카곤종, 혈관작용 소장 펩타이드 분비 종양(vasoactive intestinal peptides producing tumor, VIPoma) 등으로 분류되며 이 중 인슐린종이 60%로 가장 높은 빈도를 보인다[3,4]. 부신피질자극호르몬(adrenocorticotropic hormone)을 분비하는 췌장내분비종양은 매우 드물며, 임상적으로 부신피질자극호르몬 분비와 그에 따른 코티솔 과잉으로 쿠싱증후군을 나타낸다. 진단 당시 원격 전이를 동반하는 경우가 많아 근치적 절제가 어려우며 예후가 매우 불량한 것으로 알려져 있다[5]. 치료로는 조기에 진단된 경우 수술적 절제가 가장 효과적인 치료방법이다. 하지만 전이가 있거나 뚜렷한 종양이 발견되지 않는 경우 혈청 부신피질자극호르몬을 억제하면서 고혈압, 감염, 골다공증 등 코티솔 과잉에 의한 합병증을 조절하는 치료를 한다. 저자들은 월상안, 중심 비만, 조증(mania) 등 전형적인 쿠싱증후군으로 내원하여 부신피질자극호르몬을 분비하는 췌장에서 발생한 신경내분비종양 1예를 경험하였기에 보고하는 바이다.
증 례
55세 여자가 내원 2개월 전부터 발생한 허리 통증으로 본원 정형외과에 내원하였다. 환자는 내원 전 허리 통증에 대해 성분 미상의 주사제로 치료받은 병력이 있으며 내원 6개월 전부터 쉽게 멍이 들고 몸이 붇는 증상이 있어 쿠싱증후군에 대한 검사를 위해 내분비내과에 의뢰되었다. 고혈압, 당뇨병의 과거력은 없었으며 음주와 흡연력도 없었다. 이외에 특이한 가족력도 없었다. 내원 시 활력 증후는 혈압 130/80 mm Hg, 맥박 80/분, 호흡수 20/분, 체온 36.4°C였고 의식은 명료하였다. 키 155 cm, 몸무게 54 kg으로 체질량지수(body mass index)는 22.5 kg/m2였으며 6개월 전부터 체중증가가 있었다. 환자는 정신병의 과거력은 없었으나 가끔씩 자신도 모르게 이웃 사람들에게 물건을 사주는 행동을 하는 조증(mania)의 증상을 보였다. 양 다리의 쇠약감을 호소하였고 상하지 근위축, 월상안, 안면 홍조, 후경부 비후, 복부 자색선조 및 양손에 색소침착소견을 보였다. 복부 검진에서 복부 팽만이 있었으나 압통이나 반발통은 보이지 않았으며 간종대, 비종대도 없었다. 내원하여 시행한 혈액검사에서 백혈구 7,500/mm3 (중성구 88%, 림프구 7%), 혈색소13.6 g/dL, 혈소판 213,000/mm3였다. 생화학검사에서 포도당 229 mg/dL, 알부민 3.5 g/dL, 혈청요소질소 29 mg/dL, 크레아티닌 0.5 mg/dL, 아스파테이트 트랜스퍼라아제 18 U/L, 알라닌 트랜스퍼라아제 20 U/L, 총 빌리루빈 0.7 mg/dL였으며 알카라인 포스파타아제는 221 U/L로 증가되어 있었다. 전해질검사에서 나트륨 148 mEq/L, 칼륨 2.5 mEq/L, 중 탄산이온은 33 mmol/L로 저칼륨혈증과 대사성 알칼리혈증이 있었다. 오전 8시에 측정한 기저 혈청 코티솔 농도는 53.18 μg/dL (정상범위, 7.0-25.0 μg/dL), 부신피질자극호르몬 농도는 355 pg/mL (정상범위, 10-60 pg/mL)로 증가되어 있었다. 24시간 소변검사에서 유리 코티솔은 1,451 μg/dL (정상범위, 75-270 μg/dL)로 증가되어 있었다. 갑상선자극호르몬은 2.45 μIU/L, 유리 T4는 1.08 ng/dL로 정상범위였다. 고용량 덱사메타손 억제검사에서 덱사메타손 투여 후 혈청 코티솔 농도는 27.06 μg/dL, 24시간 소변 유리 코티솔은 2,097 μg/dL로 모두 억제되지 않았다. 뇌하수체 자기공명영상에서 종양은 관찰되지 않았다. 복부 전산화단층촬영에서 췌장 미부와 비장의 hilum을 중심으로 경계가 불명확한 저음영의 종양이 관찰되었으며 위의 소만 부위와 인접해 있었다(Fig. 1). 흉부 전산화단층촬영에서 폐 전이는 관찰되지 않았다. 림프종과의 감별을 위해 비장의 종양에 대해 초음파 유도하 조직검사를 시행하였으며 종괴 내에 다발성의 격막(septum) 혹은 hyalinized stroma 소견을 보였고 면역조직화학염색에서 신경내분비 분화소견을 보여 췌장의 신경내분비종양이 의심되었다. 환자는 비장과 췌장의 미부 절제와 위 쐐기절제술을 시행 받았으며 병리검사에서 췌장의 미부에 5 cm 크기의 경계가 불분명한 회백색의 단단한 고형의 종괴가 보였으며 유착된 비장의 실질 및 위의 근육층으로 침범이 관찰되었다(Fig. 2A). 현미경검사에서 종양세포는 크기가 유사한 둥근 세포들로 구성되어 있었으며 중등도의 세포질 및 salt-and-pepper 양상의 염색질을 보이고 있었고 유사분열은 2/10 high power field 미만이었다. 종양세포들은 기둥 혹은 이랑 모양의 군집을 이루며 혈관섬유조직 사이에 분포하고 있었다(Fig. 2B). 종양 내에 괴사와 림프관 및 신경 주위 침습이 있었으며 종양세포들은 췌장의 피막을 뚫고 비장 실질과 유착된 위의 점막하 조직으로 침윤하고 있었다. 면역조직화학염색에서 종양세포는 신경내분비 표지자인 synaptophysin과 CD56 및 부신피질자극호르몬에 양성으로 염색되어(Fig. 2C) 분화도가 좋은 췌장내분비샘암종으로 진단하였다. 췌장 주변 및 위 주변 림프절 중 2개에서 내분비샘암종의 전이가 관찰되었다.
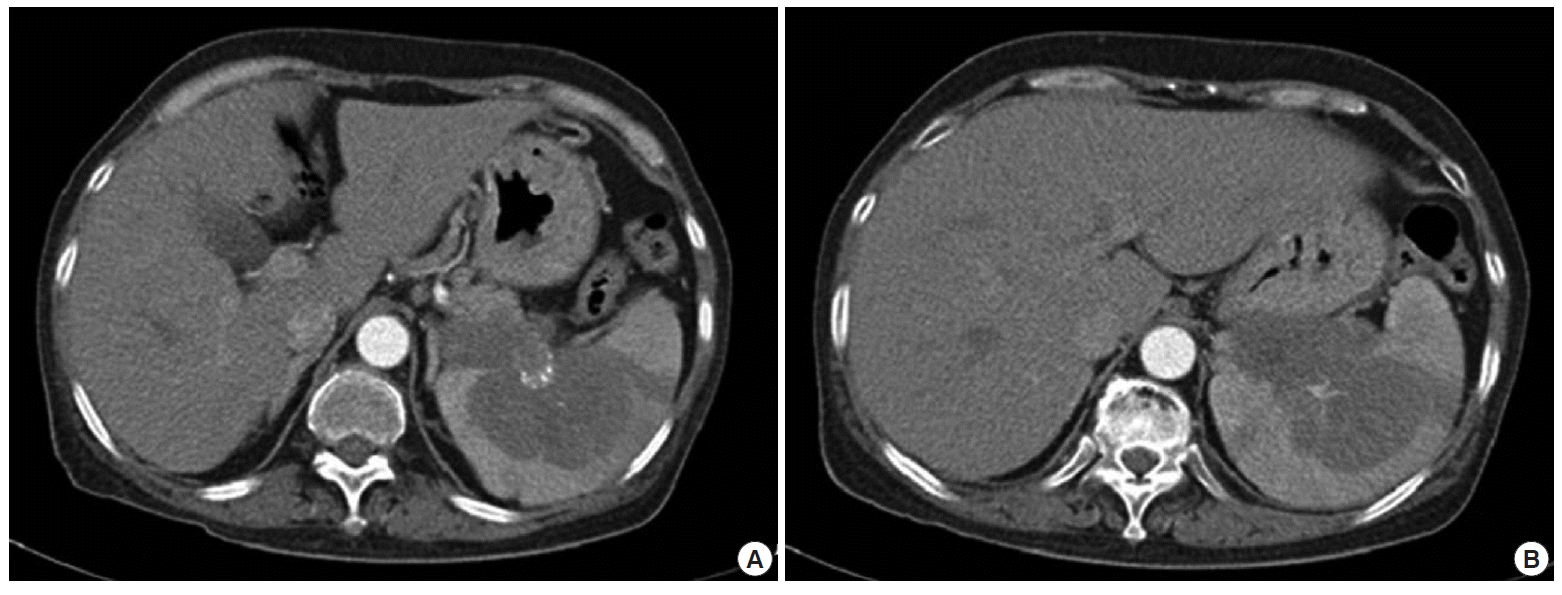
Contrast enhanced abdomen computed tomography shows a large ill-defined low attenuation mass in pancreatic tail, and spleen (A), abutting stomach (B).
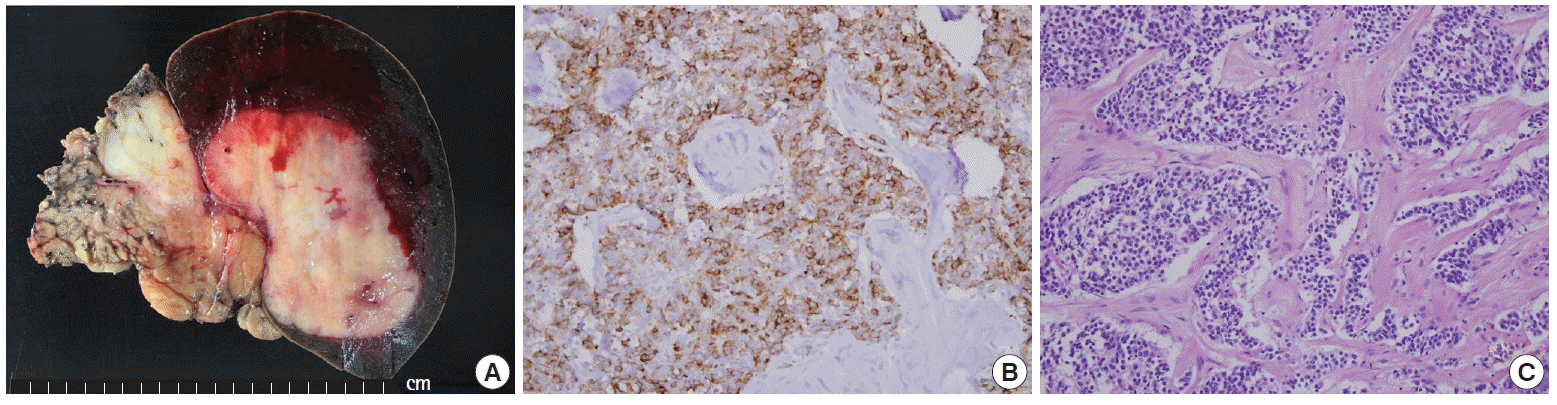
There is an ill-defined white to gray solid mass with necrosis (5 cm in diameter) in the pancreatic tail. (A) The tumor invades into the splenic parenchyma and muscle layer of the adhered stomach. (B) On microscopic exam, the tumor shows a proliferation of round, uniform cells with a moderate amount of cytoplasm and salt-and-pepper chromatin. They are arranged in distinct nests with trabecular and gyri patterns and separated by fibrovascular stroma (H&E, × 200). (C) On immunohistochemical stains, the tumor cells are positive for adrenocorticotropic hormone (immunohistochemical stain, × 200).
수술 2주 후 시행한 혈액검사에서 혈청 코티솔은 15 μg/dL, 부신피질자극호르몬은 75 pg/mL로 감소하였고 24시간 소변검사에서 유리 코티솔은 76 μg/dL로 감소하였으며, 환자의 근무력, 불안, 조증 등의 증상은 호전되었다. 이후 환자는 특별한 증상 없이 외래 경과관찰하던 중 수술 후 1년 4개월에 시행한 추적 복부 전산화단층촬영에서 다발성 간 전이가 발견되었으며 부신피질자극호르몬 농도가 156 pg/mL까지 상승하는 소견을 보여 간동맥화학색전술(transhepatic arterial chemoembolization)을 시행하였으며 시술 후 부신피질자극호르몬 농도는 75 pg/mL까지 감소하였으나(Fig. 3) 복부 전산화단층촬영에서는 안정병변(stable disease) 소견을 보였다. 현재 한 달에 한 번씩 octreotide 30 mg 근육주사 투여하면서 경과관찰 중이다.
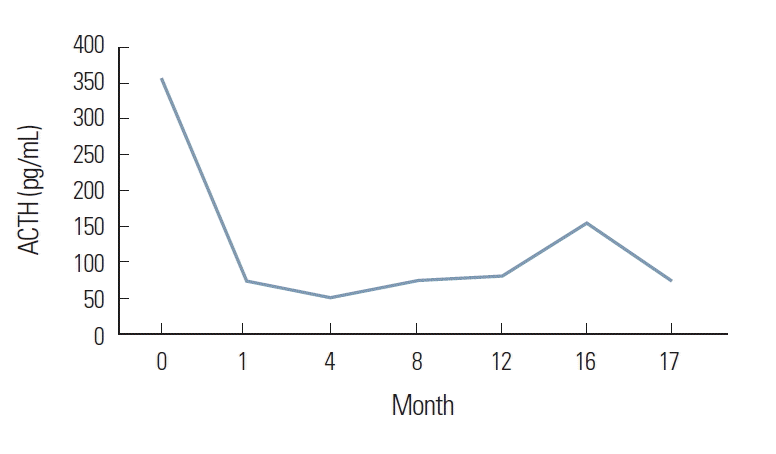
Graph shows serum adrenocorticotropic hormone level change over time. Month 0: at diagnosis. Month 1: post operation. Month 16: on recurrence. Month 17: after transarterial chemoembolization.
고 찰
췌장의 신경내분비종양은 전체 췌장암의 1-2%를 차지하는 드문 종양이며 그 중 부신피질자극호르몬을 분비하는 췌장의 신경내분비종양은 전 세계적으로 약 110예 가량 보고된 매우 드문 종양이다[3]. 임상적으로 이소성 부신피질자극호르몬의 과분비에 의한 코티솔 과다로 쿠싱증후군을 나타낸다. 이소성 쿠싱증후군을 일으키는 대표적인 종양은 기관지 카시노이드종양, 흉선암, 췌장내분비종양, 갑상선수질암, 크롬친화세포종이며 약 10% 정도가 ACTHoma에 기인한다. 한 연구에 의하면 이소성 쿠싱증후군의 경우 저칼륨혈증(70%)과 코티솔 과잉에 의한 정신 증상(50%)이 흔하게 나타나며 중증 감염(15%)과 골다공증, 골절(50%)도 흔하게 나타나는 특징이 있다[6].
본 환자는 월상안, 후경부 비후, 조증 등의 쿠싱 증상을 보였으며 혈액검사에서 저칼륨혈증과 대사성 알칼리증을 보였다. 혈장 부신피질자극호르몬과 소변 유리 코티솔 농도가 증가되어 있어 부신피질자극호르몬 의존성 쿠싱증후군을 의심할 수 있었다. 뇌 자기공명검사에서 뇌하수체에 이상소견은 관찰되지 않았으며 고용량 덱사메타손 억제검사에서 혈청과 소변 코티솔이 억제되지 않아 이소성 부신피질자극호르몬의 분비에 의한 쿠싱증후군이 의심되었다. 복부 전산화단층촬영에서 췌장 미부와 비장에 걸쳐 있는 종양이 관찰되었으며 이에 대한 조직검사에서 신경내분비종양의 소견을 보였고 면역염색에서 부신피질자극호르몬에 양성소견을 보여 ACTHoma로 진단되었다. 또한 수술적 제거 후 혈청 코티솔과 부신피질자극호르몬이 감소하고 쿠싱증후군의 호전소견을 보여 췌장의 신경내분비종양에 의한 이소성 쿠싱증후군으로 확진할 수 있었다.
ACTHoma는 다른 신경내분비종양에 비해 전이가 흔하며 95% 이상에서 전이를 보이는 악성도가 높은 암으로 환자의 60%가 2년 내에 사망하고 5년 생존율이 16%로 예후가 매우 불량하다[3,7]. 신경내분비종양이 간 전이를 동반한 경우 5년 생존율이 50% 이하로 나빠지며 환자 삶의 질이 나빠지게 된다. 신경내분비종양의 치료는 수술적 치료가 가장 좋은 치료이나 ACTHoma의 경우 진단 당시 전이를 동반하는 경우가 많아 근치적 절제가 어렵고 전이를 동반하지 않는 경우에도 수술적 치료성적은 좋지 않다. 한 연구에 따르면 간 전이가 있는 10명을 포함한 총 12명의 ACTHoma 환자를 대상으로 수술적 치료를 하였을 때 2.5년에 50%가 사망하였으며 생화학적 완치가 된 환자는 한 명도 없었다[8]. 본 환자는 수술 후 혈청 부신피질자극호르몬, 코티솔 농도가 정상으로 감소하였으나 수술 후 1년 4개월 후 간 전이가 진단되었을 당시 156 pg/m으로 증가되었다.
간 전이를 동반한 신경내분비종양에 대한 정립된 치료방법은 아직 없다. 간 이식, 간절제술 등 수술적 치료는 생존률을 향상시키고 증상 호전을 기대할 수 있으나 완치는 드물다. 간 전이를 동반한 췌장내분비종양의 경우 동맥화학색전술은 78-84%의 반응률을 보인다[3]. 약물치료로는 streptozocin 기반의 전통적인 항암화학요법과 octrotide 같은 소마토스타딘 유사체를 사용할 수 있다. 최근에는 vascular endothelial growth factor의 경로를 차단하는 티로신 키나아제 억제제인 sunitinib과 mammalian target of rapamycin 저해제인 everolimus 등의 경구약제도 사용할 수 있게 되었다. 105명의 진행된 췌장내분비종양 환자를 대상으로 한 무작위 임상시험에서 streptozocin과 doxorubicin 병합요법을 사용하였을 때 69%의 반응률을 보였으며 2.2년의 평균 생존율을 보였다[9]. 분화도가 좋은 전이성 췌장내분비종양 171명의 환자를 대상으로 한 연구에서 sunitinb를 투여한 그룹의 평균 무진행생존율(progression free survival)이 대조군에 비해 유의하게 높았다[10]. 진행된 410명의 췌장내분비종양을 대상으로 한 everolimus의 효과에 대한 연구에서 everolimus 단독투여 군이 대조군에 비해 평균 무진행생존율이 유의하게 높았다[11]. 그러나 streptozocin 기반의 함암화학요법은 심한 골수부전, 신부전 등의 합병증이 많은 환자에서 나타나 사용이 제한적이며 sunitinib, everolimus 등의 분자표적치료는 고가이며 우리나라에서 보험 적용이 안 되는 문제점이 있다. Octreotide는 기능성 췌장내분비종양에서 소마토스타틴 수용체를 가지는 경우 증상 조절에 효과적이며 카시노이드종양, 글루카곤종양, 혈관작용 소장 펩타이드 분비 종양에서 효과적인 것으로 알려져 있다. 또한 몇몇 연구에서 octreotide가 진행된 카시노이드종양에 대해 종양 크기를 줄이는 효과가 있다는 것이 알려졌다[12,13]. 반면 ACTHoma에 있어서 octreotide에 대한 반응이 미흡하다는 보고가 있으며[3] 현재 octreotide가 췌장의 신경내분비종양에 대해 크기 감소효과가 있는지는 불명확하다. 따라서 진행된 ACTHoma의 경우 쿠싱증후군에 의한 증상 완화를 위해 양측 부신절제술을 시행하거나 ketoconazole이나 metyrapone을 사용하여 코티솔 합성을 억제시키는 내과적 치료를 할 수 있다. 본 환자는 수술적 치료 후 쿠싱증후군의 호전을 보였으며 부신피질자극호르몬, 코티솔 수치도 정상화되었다. 수술 후 1년 4개월째에 간 전이를 보였으나 월상안, 후경부 비후, 기존 허리통증의 악화, 조증 등 쿠싱증후군의 재발을 보이지는 않아 부신절제술이나 ketoconazole 등의 약제를 사용하지 않았으며, 간 전이에 대해서는 간동맥화학색전술을 시행하였고 octreotide를 사용하면서 경과관찰 중이다. 본 증례는 허리통증, 전신쇠약 등으로 내원한 환자에서 부신피질자극호르몬을 분비하는 췌장내분비종양에 의한 이소성 쿠싱증후군을 진단하고 수술적 치료와 재발에 대해 간동맥화학색전술로 종양과 증상 조절을 한 증례이다. 향후 골다공증, 감염 등 쿠싱증후군의 합병증에 대한 예방 및 치료를 하면서 간 전이가 재발 시 반복적 간동맥화학색전술을 시행하고, 만약 타 장기로의 전이 시 항암화학요법치료를 고려할 수 있을 것으로 생각된다.
Acknowledgements
본 연구는 순천향대학교 학술연구비의 일부 지원으로 수행하였다.