INTRODUCTION
Inflammatory demyelinating central nervous system (CNS) diseases as diverse as neuromyelitis optica (NMO), multiple sclerosis (MS), transverse myelitis, acute disseminated encephalomyelitis (ADEM), or optic neuritis (ON) have shared clinical, radiological, and immunological features, particularly at onset [1,2]. Many demyelinating disease-associated antibodies have been described [3,4] as important diagnostic and prognostic biomarkers. Recently, antibodies against myelin oligodendrocyte glycoprotein (MOG) have gained attention. High titers of anti-MOG antibodies are reported to be predominant in pediatric patients with acquired inflammatory demyelinating diseases of the CNS such as recurrent ONs, ADEM followed by ON, and multiphasic disseminated encephalomyelitis [2,5–7]. Patients with anti-MOG antibodies appear to have a unique clinical, radiological, and therapeutic profile. Therefore, new specific therapeutic strategies for anti-MOG antibody-associated disease are under development. Because few cases of MOG seropositivity have been reported in Asian children [8,9], we here describe a Korean child with recurrent unilateral ONs and a single ADEM episode who showed high titers of MOG antibodies. This study was approved by the institutional review board of the Soonchunhyang University Bucheon Hospital with informed consent (2019-11-041).
CASE REPORT
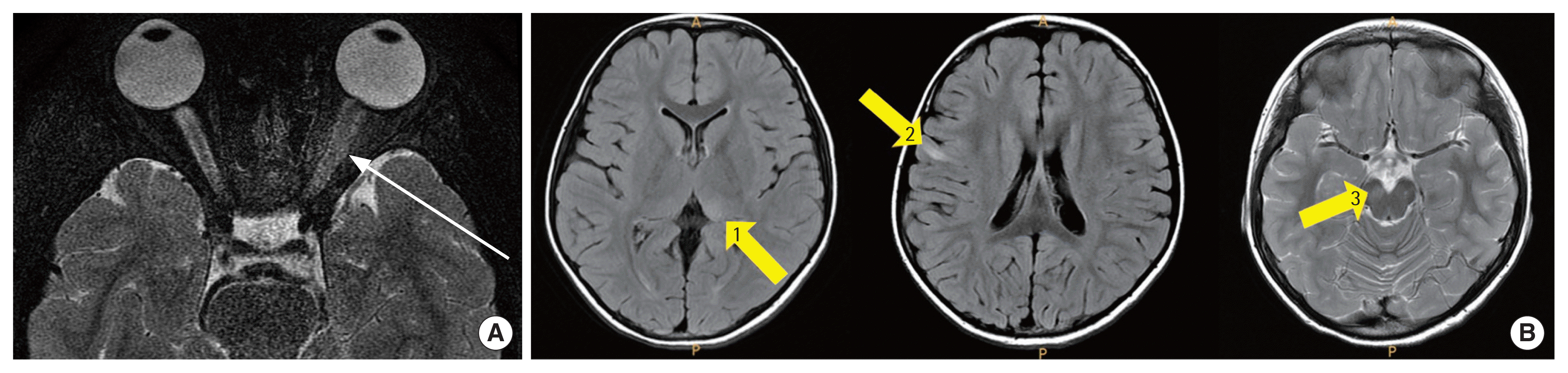
A 6-year-old Korean girl with sudden loss of vision in her left eye visited Soonchunhyang University Bucheon Hospital. She had been healthy previously and had an unremarkable medical and family history. The ophthalmologic examination revealed a decreased visual acuity, loss of color vision, and the fundoscopy showed a left optic disc swelling. Brain and orbit magnetic resonance imaging (MRI) revealed enlarged left optic nerve with perineural fat infiltration on T2-weighted images (T2-WIs) (Fig. 1A), as well as increased signal intensities on T2-WIs and fluid-attenuated inversion recovery images in the bilateral frontal subcortical white matter, left thalamus, and right cerebral peduncle; notably, there were no neurological symptoms (Fig. 1B). The lesions had subtle signal changes in diffusion images and were not enhanced with gadolinium. Her cerebrospinal fluid (CSF) showed lymphocytosis, but protein and sugar were in the normal range (Table 1). No organisms were found in her serum or CSF; oligoclonal bands (OCBs) and anti-aquaporin-4 antibodies (AQP4 abs) were negative. After high-dose intravenous (IV) methylprednisolone for 5 days followed by a tapering course of oral steroids for 8 weeks, her visual acuity improved dramatically from finger counting only to 0.6 after 1 month after the 1st episode. Maintenance treatment for acquired demyelinating syndrome was not started at that time; however, due to this clinically isolated syndrome with subclinical brain MRI lesions, she visited the outpatient clinic regularly under the strong suspicion of a relapsing course of an acquired demyelinating disease. One month after cessation of the oral steroids, she complained of drowsiness, poor appetite, and gait disturbances with mild fever for several days. On her second brain MRI, she presented newly developed, extensive, and ill-defined hyperintensities on T2-WIs. These lesions were detected in the subcortical white matter around the 4th ventricle, deep gray matter, both multifocal cortexes, left cerebral peduncle, and right thalamus. Neither the previous hyperintense lesions nor a swelling of the optic nerve were detectable (Fig. 2). Additionally, spinal MRI T2 images showed newly apparent ill-defined hyperintense lesions at the C3 level. After steroid pulse therapy, she nearly fully recovered within 3 days. The clinical symptoms, the brain MRI, and the CSF tests satisfied the diagnostic criteria for ADEM; considering her previous episode of steroid-responsive unilateral ON, regular subcutaneous injections of interferon (IFN) β-1a were started, accompanied by oral steroid tapering. At the 2nd episode her visual acuity improved to 0.9 on the left side.
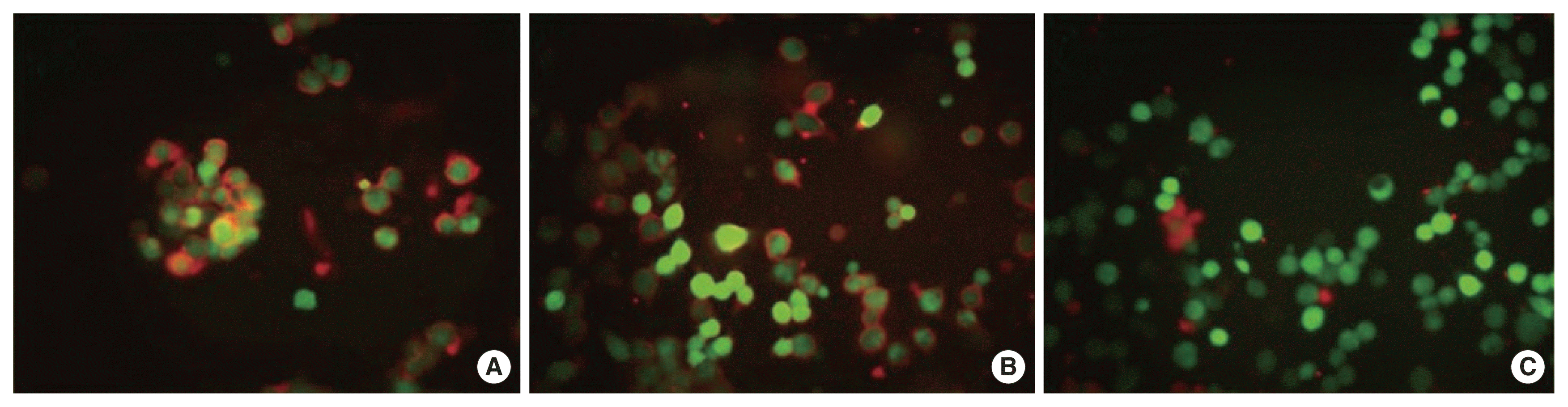
Despite regular maintenance treatment with IFN β-1a, she experienced a 3rd episode, 3 months later, as a unilateral right ON. This time, the brain and spinal MRIs showed no newly developed lesion, and the extensive previous lesions had disappeared. After high-dose steroid injection for 5 days, the visual acuity of the right eye improved gradually from 0.2 to 0.6. Throughout these three episodes, serological tests associated with autoimmune diseases, viral markers, anti-AQP4 abs, and CSF OCBs were all negative, with the immunoglobulin G index in the normal range (Table 1). Finally, a serological examination for anti-MOG antibodies was performed, and a high titer of serum anti-MOG antibody was detected (Fig. 3). As a long-term treatment of MOG autoantibody-associated demyelinating diseases, regular IV immunoglobulin or mycophenolate mofetil was considered as a treatment option in this patient.
DISCUSSION
In this patient, we decided to test anti-MOG antibody considering the following facts: recurrent unilateral ON at a young age, mainly with ADEM-like and deep gray matter lesions in the brain MRI; good response to steroids; relapse despite IFN β-1a treatment; and anti-AQP4 ab-negative findings in serologic analysis. Recently, MOG antibodies have been extensively studied in relapsing courses of demyelinating syndromes, especially in pediatric cases. They sometimes meet the existing MS and NMO spectrum disorder (NMOSD) criteria, but exhibit different clinical courses, which is reflected in the revised criteria [10]. The treatment strategy for MOG antibody-positive diseases has not yet been fully established, and the evidence for an optimal approach is undergoing collection. IV methylprednisolone is considered an effective drug in the acute phase and IV immunoglobulin was reported to be helpful in prevention of relapse [6]. Corticosteroids, mycophenolate mofetil, and methotrexate have all been used as disease-modifying drugs, while immunomodulating drugs for MS, such as IFN, have typically been ineffective [1,6]. We recommend performing a MOG antibody test in children who exhibit suspected atypical MS or relapsing anti-AQP4 ab-negative NMOSD. This establishes the correct diagnosis and enables proper long-term treatment.