Tatton-Brown-Rahman Syndrome: A Report of a Case with a Novel DNMT3A Mutation Presented Hemihypertrophy
Article information
Abstract
Tatton-Brown-Rahman syndrome (TBRS) is a relatively new congenital anomaly syndrome manifesting overgrowth and a broad spectrum of intellectual disability. It is caused by pathogenic variants in the DNA methyltransferase 3 alpha (DNMT3A) gene, mainly de novo inheritance. Overgrowth, mild-to-severe intellectual disability, and other clinical features of TBRS may affect the quality of life of patients and their family members. Thus, early diagnosis by genetic testing and management of these symptoms is critical. We report a case of a 17-year-old male patient with hemihypertrophy who suffered back pain since school age, diagnosed with TBRS-identified DNMT3A gene mutation.
INTRODUCTION
Human overgrowth syndromes are conditions with height and/or head circumference ≥2 standard deviations above the mean. Overgrowth and intellectual disability (OGID) syndrome is when additional phenotypic abnormalities, most commonly intellectual disability associated due to single-gene disorders [1,2]. Tatton-Brown-Rahman syndrome (TBRS) is a relatively new congenital overgrowth syndrome first described in 2014, with approximately 80 reported thus far, and first reported in 2018 in Korea [3]. TBRS is known to be caused by a mutation in the DNA methyltransferase 3 alpha (DNMT3A) gene inherited in an autosomal dominant manner, but mainly de novo inheritance [2,4]. The most common clinical features from previously reported TBRS include overgrowths such as macrocephaly, a broad spectrum of intellectual disabilities with or without autism spectrum disorder, and dysmorphic facial features. Also, upper back curving, heart defects, flat feet, weak muscle tone, and flexible joints may be seen [4]. TBRS’s distinctive facial features include low-set, horizontal eyebrows, prominent upper central incisors, rounded faces, and narrow palpebral fissures; however, facial characteristics alone cannot distinguish from other OGID syndrome because it is less specific compared to other clinical features [5]. Thus, multigene panel sequencing is crucial to confirm and differentiate TBRS from other overgrowth syndromes.
CASE REPORT
Our patient was born to non-consanguineous Korean parents at full-term with 3,089 g of weight (75th–90th percentile) by vaginal delivery. Our neurosurgery and orthopedics department referred him to our clinic for hemihypertrophy at age 17 years old. The patient has no prior medical or trauma history but has experienced lower back pain for as long as he can remember. The symptoms worsened 1 year ago when he started high school and spent more time sitting in classes. Nevertheless, he had average intellectual abilities to keep up with his classes. During the physical examination, no notable facial features were observed, except for being overweight (height: 179 cm, weight: 88.2 kg, body mass index: 27.5 kg/m2), and having a significantly larger left hand and foot compared to the right side. There was about a 3 cm discrepancy in the level of both femoral heads and mild lumbar scoliosis from the whole spine X-ray (Fig. 1A, B). To differential molecular diagnosis of hemihypertrophy, we performed diagnostic exome sequencing tests related to overgrowth and vertebral anomaly. In the c.2074C>T region of the DNMT3A gene, we identified a heterozygous nonsense variant mutation, which is commonly associated with Tatton-Brown-Rahman syndrome (TBRS) (Fig. 2). The pathogenicity of the variant was confirmed, and it was classified as a de novo mutation based on Sanger sequencing results obtained from the patient’s parents. Clinical features of TBRS include overgrowth and curvature of the spine, which explain the patient’s chronic back pain. Further investigations were performed to assess other clinical symptoms related to TBRS, such as blood laboratory tests, echocardiogram, and abdominal and pelvic computed tomography (CT). No other significant findings were observed, including intellectual disability. The patient has been referred to pediatric orthopedics for monitoring the progression of back pain and consultation regarding rehabilitation options and the potential need for extension surgery.
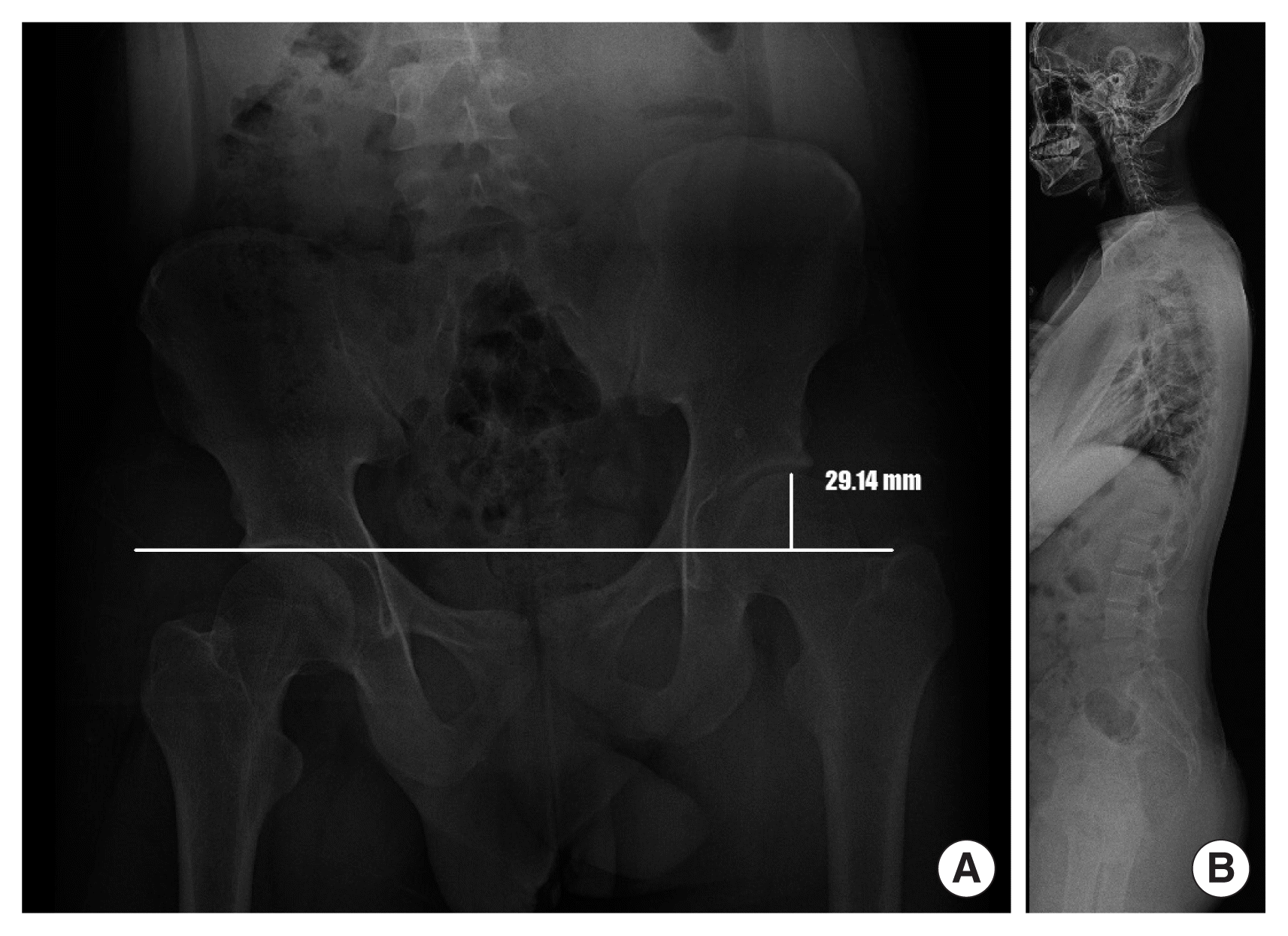
The patient’ whole spine anteroposterior (A) and lateral (B) X-ray also showed arterial convexity to the right side, with about a 3 cm discrepancy to the left in the level of both femoral heads.
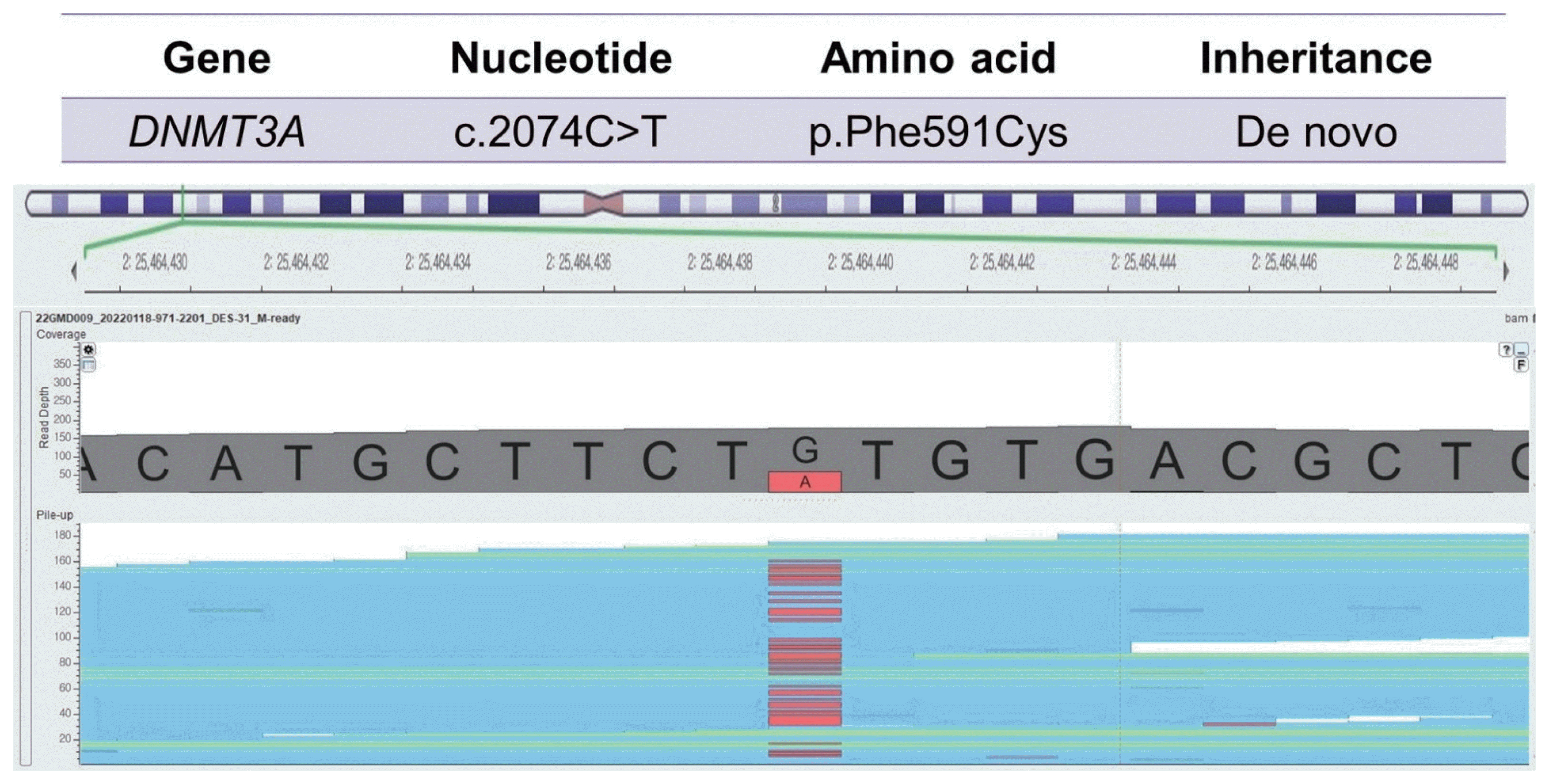
Sanger sequencing demonstrated a variation (c.2074C>T), resulting in a mutation of the DNA methyltransferase 3 alpha (DNMT3A) gene.
This report was proved by the institutional review board (IRB) for the publication of clinical details and images (IRB no., 2023-05-012).
DISCUSSION
TBRS is a relatively new congenital anomaly syndrome manifesting overgrowth and possibly intellectual disability. The function of the DNMT3A gene has yet to be fully understood. However, because the enzyme DNMT3A is responsible for de novo methylation, it is hypothesized to decrease methylation (hypomethylation), resulting in transcriptional repression [4]. Often, TBRS patients are misdiagnosed with other OGID syndromes, such as Sotos syndrome and Weaver-Smith syndrome, and TBRS was misdiagnosed as Sotos syndrome until TBRS was first reported in 2014. Sotos syndrome is the most common syndrome in OGID syndrome caused by pathogenic variants in the NSD1 gene, and Weaver-Smith syndrome is caused by a variant in the EZH2 gene. Unlike Sotos syndrome and Weaver syndrome, TBRS has a higher reported rate of congenital heart defects and acute myeloid leukemia [2].
Surveillance in TBRS includes monitoring behavior or developmental progress, neurologic manifestations such as seizures, sleep apneas, cardiac anomalies, hematologic malignancies, and genetic counseling [4,6]. Management includes supportive care based on symptoms, rehabilitation for developmental delay or kyphoscoliosis, encouraging patients to keep up with school work, and supporting their emotions.
TBRS should be considered if generalized overgrowth and mild-to-severe developmental delay or intellectual disability are shown in pediatric patients [7]. Unlike other overgrowth syndromes that are inherited disorders, such as Marfan syndrome, although Marfan syndrome rarely causes intellectual disability, TBRS is usually caused by a de novo pathogenic variant [5]. Most TBRS cases were first spotted concerning intellectual disability with or without overgrowth and distinct facial features. So far, two cases of TBRS have been reported in Korea, and they both had mild to moderate intellectual disabilities with subtle dysmorphic facial features [3,8]. Lee et al. [8] reported the first Korean family with TBRS, and both the 11-year-old patient and her mother had mild developmental delay, obesity, and macrocephaly with dysmorphic facial features, including a round face, horizontal eyebrows, and narrow palpebral fissures without spine abnormality. The study by Shen et al. [6] with three TBRS patients had intellectual disabilities, overgrowth and but no spine abnormality. However, our patient was referred for unknown back pain and hemihypertrophy with average intellect and no recognizable facial features, which do not correlate with previously reported TBRS. Before TBRS was first reported, it was easily missed or misdiagnosed due to subtle dysmorphic features compared with other OGID syndromes [2]. Therefore, targeted multigene panel sequencing, including DNMT3A or exome sequencing, maybe a helpful diagnostic tool for early diagnosis and surveillance in pediatric patients with overgrowth with or without intellectual disability.
Notes
No potential conflict of interest relevant to this article was reported.