A Case of Renal-limited Lupus-like Nephritis
Article information
Abstract
Systemic lupus erythematosus (SLE) is a systemic autoimmune disease that is clinically heterogeneous and affects multiple organs particularly the kidney. Lupus nephritis is a common and severe manifestation of SLE in which immune-mediated inflammation can lead to permanent damage within the kidney, resulting in end stage renal failure. Recently a renal biopsy showed lupus nephritis on a 40-year old female without any other features of SLE such as clinical symptoms and autoantibodies including antinuclear antibody and anti-dsDNA. The renal biopsy showed that histopathological change of global and segmental sclerosis of glomeluri, diffuse proliferative nephritis with crescent formation compatible with class IV lupus nephritis. She was treated with systemic corticosteroids and pulse cyclophosphamide, followed by mycofenolate mofetil. During two years of follow-up, there have been no clinical or laboratory findings to meet the diagnostic criteria of SLE, suggesting that isolated lupus nephritis could occur without SLE.
INTRODUCTION
Systemic lupus erythematosus (SLE) is a systemic autoimmune disease that is characterized by the production of autoantibodies to nuclear and cytoplasmic antigens and could affect multiple organ systems including kidney [1]. In particular, renal involvement such as lupus nephritis (LN) is a common manifestation in SLE in which autoantibodies are thought to participate in the formation of glomerular immune complexes, initiating the inflammatory responses [2,3]. Renal involvement in SLE is still one of the strongest predictors for morbidity and mortality. Depending on the studied population, LN occurs in 43% to 55% of SLE patients and usually within the first year of disease. In some aggressive cases such as class IV LN, up to 25% of LN patients develop end-stage renal disease (ESRD) 10 years after onset of renal disease [4,5].
Antinuclear antibody (ANA) elevated in the serum form the mainstay of serologic testing for the diagnosis of SLE, having a frequency of 95% in patients with SLE [6,7]. Nevertheless, cases of SLE with a negative ANA have been reported, meaning that a negative ANA in serology could not rule out SLE [8,9]. We report a case with isolated LN characterized by pathological findings without other clinical and serologic abnormalities compatible with the diagnosis of SLE.
CASE REPORT
A 40-year-old female with no family history of renal or autoimmune disease was noted to have proteinuria during a routine examination which was performed as part of prenatal screening program four years ago. At this point, the patient decided to not receive treatment at that time. Recently, she was found to have proteinuria again during an annual health check. Her blood pressure was 120/80 mm Hg, the pulse rate 78/min, the respiratory rate 20/min and the body temperature 36.5°C. She had white blood cell count of 8,180/mm3 (range, 3,700 to 9,000/mm3), hemoglobin level of 11.8 g/dL (range, 11 to 16 g/dL), platelet count of 293,000/mm3 (range, 130,000 to 370,000/mm3), serum blood urea nitrogen 13.4 mg/dL (range, 8 to 23 mg/dL), creatinine of 0.9 mg/dL (range, 0.6 to 1.2 mg/dL), serum protein 4.8 g/dL (range, 5.8 to 8.0 g/dL), serum albumin 3.1 g/dL (range, 3.1 to 5.2 g/dL), serum total cholesterol 242 mg/dL (range, 115 to 230 mg/dL), serum triglyceride 158 mg/dL (range, 50 to 200 mg/dL), serum high density lipoprotein cholesterol 45.3 mg/dL (range, 35 to 65 mg/dL), serum low density lipoprotein cholesterol 173 mg/dL (range, 77 to 135 mg/dL), and 24-hour urine protein of 6,624 mg. Her spot urine urinalysis revealed urine red blood cell 5–9/high power field (HPF) (0–1/HPF), urine protein 3+. She also had the erythrocyte sedimentation rate 16 mm/hr (range, 1 to 20 mm/hr), and C-reactive protein 0.02 mg/dL (range, 0 to 0.3 mg/dL). C3 and C4 levels were normal. ANA, anti-dsDNA antibody, anti-cardiolipin antibody, anti-beta2 GP1 antibody, lupus anticoagulant, anti-Sm antibody, anti-Ro/La antibody, anti-RNP antibody, and rheumatoid factor were all negative. HIV and hepatitis B and C viruses serologies were negative. She had no signs or symptoms of SLE. Physical examination was unremarkable. Renal biopsy revealed segmental endocapillary proliferative glomerulonephritis with a marked global thickening of the capillary walls, which can be found in LN class IV-S (A/C) (Fig. 1). Activity score was 6, and chronicity score was 5. Immunofluorescenc studies demonstrated global granular immunoglobulin G (IgG), immunoglobulin M (IgM), immunoglobulin A (IgA), C3, C4, and C1q deposits in glomerular capillary walls and in the mesangium (Fig. 2). Electron microscopy showed global mesangial and subendothelial electron-dense deposits (Fig. 3).
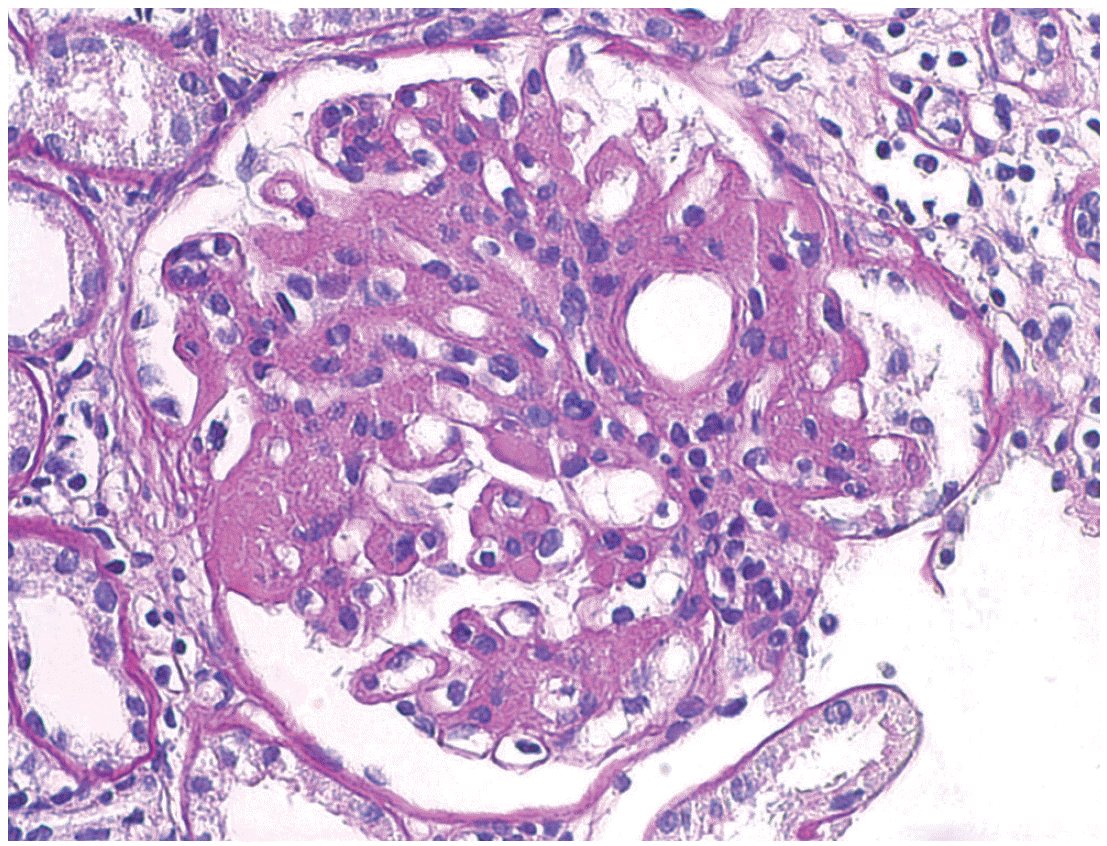
The glomerulus shows segmental endocapillary hypercellularity and marked global thickening of the capillary walls with eosinophilic subendothelial deposits (periodic-acid-Schiff stain, × 400).
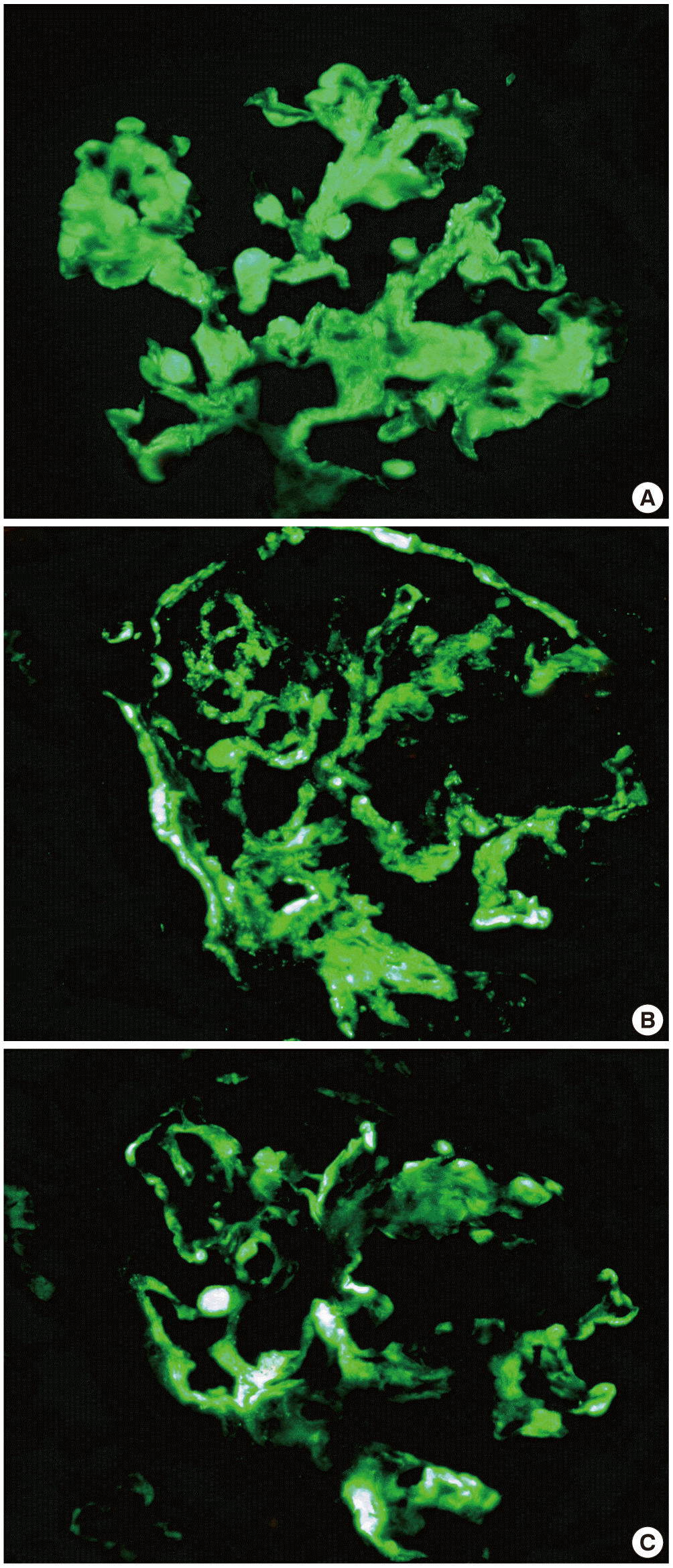
(A) Immunoglobulin G (IgG), (B) C3, (C) C4. Immunofluorescence microscopy reveals global granular IgG, immunoglobulin M, immunoglobulin A, C3, C4, and C1q deposits in glomerular capillary walls and in the mesangium (× 400).
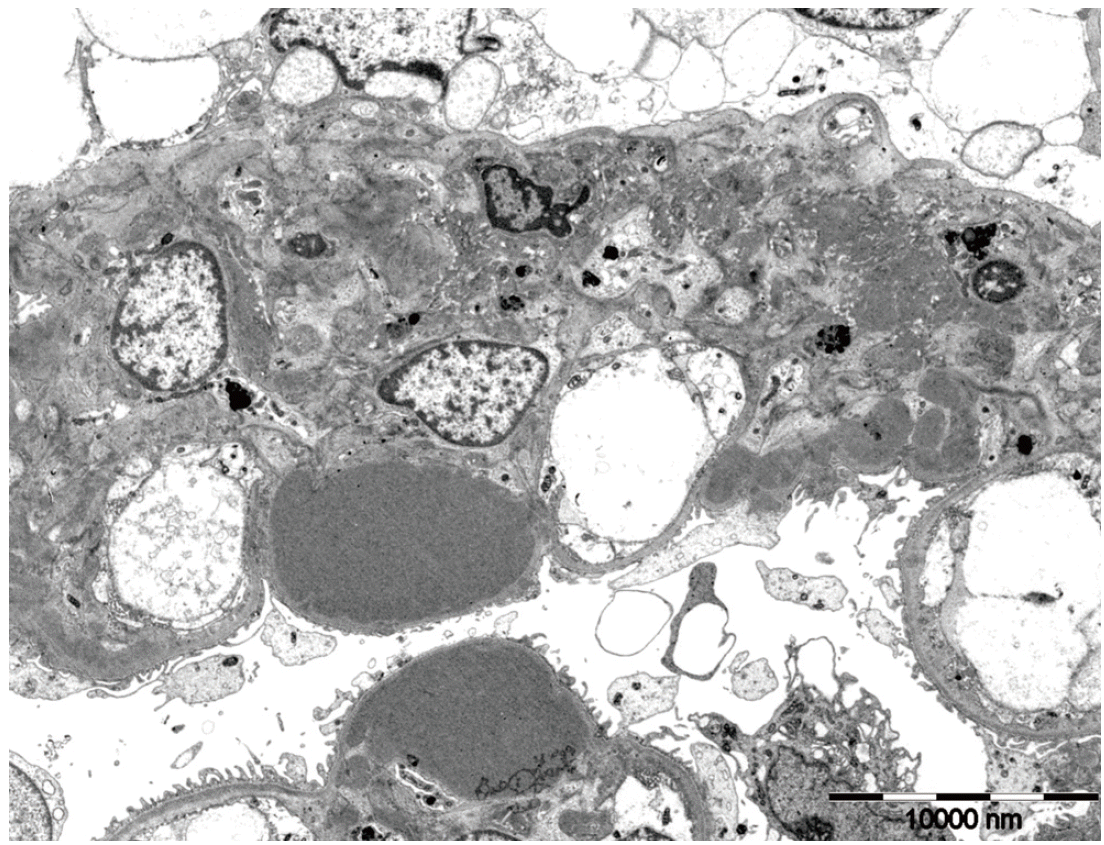
The electron micrograph shows abundant subendothelial electron dense deposits together with mesangial deposits (× 3,000).
The patient was given three monthly Intravenous cyclophosphamide (500 mg once a month) treatments in conjunction with pulse IV methylprednisone (62.5 mg once a month), followed by mycofenolate mofetil (MMF; range, 2 to 3 g/day) and prednisolone as maintenance therapy. Her proteinuria has declined up to 43 mg/day and her renal function continued to be normal and her serum albumin elevated to 3.8 g/dL. A repeat ANA and other autoantibodies including anti-dsDNA remain negative. The patient is currently on MMF and has not developed signs or symptoms of SLE.
DISCUSSION
The diagnosis of SLE is based on clinical and laboratory criteria developed by the American College of Rheumatology. A person has SLE if any 4 out of 11 criteria are present simultaneously or serially on two separate occasions [10]. However, a diagnosis of SLE can be made in a patient having fewer than four of these symptoms. In fact, small groups of patients with SLE have been reported to have negative ANA and negative anti-DNA antibody. Even if a single kidney biopsy finding does not fulfill the accepted diagnostic criteria of SLE, the specific pathological features in LN could be considered to make a diagnosis of this as ‘isolated LN’ [9]. The exact underlying mechanisms in the pathogenesis of LN are still not well understood. LN has been considered as a classical immune complex-induced glomerulonephritis due to deposition of preformed circulating immune complexes that stain dominantly for IgG or binding of autoantibodies to glomerular antigens [2]. A diffuse proliferative LN (class IV) is both the most common and the most severe subtype. In this LN, more than 50% of glomeruli are involved which can be segmental or global, and active or chronic, with endocapillary or extracapillary proliferative lesions. Using electron microscopy, subendothelial deposits are noted, and some mesangial changes may be present. Immunofluorescence reveals the so-called ‘full house’ stain, staining positively for IgG, IgA, IgM, C3, and C1q. Clinically, hematuria and proteinuria is present, frequently with nephrotic syndrome, hypertension, hypocomplementemia, elevated anti-dsDNA titers, and elevated serum creatinine [3].
Our patient had proteinuria, hypertension, or other findings of SLE including both negative ANA and anti-dsDNA antibody as well as normal serum levels of C3 and C4. She had an IgG-dominant immune-complex-mediated glomerulonephritis with fullhouse findings such as simultaneous deposits of IgA, IgM, C1q, and C3 in renal biopsy specimens. She also had tubuloreticular inclusions and global mesangial and subendothelial electron-dense deposits. These biopsy findings were highly suggestive of LN. During the follow-up period, she did not fulfill the diagnostic criteria of SLE and also the repeat ANA anti-dsDNA in serum showed all normal levels. In contrast, Huerta et al. [9] reported four female adult cases with renal biopsy findings highly suggestive of LN but without signs, symptoms or serologies of SLE. During the period of follow-up, despite aggressive therapies including steroids, MMF and/or cyclophosphamide, three of the four patients progressively led to ESRD and the other one proceeded to chronic kidney disease. In another study, Baskin, et al. [8] described a child who had negative lupus serology and full-house nephropathy in renal biopsy findings. During the subsequent 17 months, her renal function continued to deteriorate (glomerular filtration rate 17.9 mL/min/1.73 m2).
Most cases have been reported in children who showed a lupuslike nephritis at the time of biopsy but finally developed clinical and laboratory findings of SLE. In particular, the patients with LN V (membranous) and LN II (mesangial proliferative) tended to develop over SLE later.
In conclusion, this patient has isolated LN compatible with specific pathological characteristics of a diffuse proliferative glomerulonephritis (LN IV). During the follow-up period of 36 months, the patient developed no extra-renal features or serological findings of SLE, with maintenance of normal renal function and partial response to the treatment unlike the previous poor prognostic reports, which suggest that LN may be developed in the setting of negative ANA and/or anti-dsDNA.