INTRODUCTION
Neonatal diabetes mellitus (NDM) is defined as severe hyperglycemia that usually develops before 6 months of life and requires treatment [1–3]. NDM is a rare neonatal disease, with an incidence of approximately one in 90,000–160,000 infants worldwide [1]. Neonatal hyperglycemia is common on days 3 to 5 after birth, but it is unusual for it to persist beyond 10 days. Premature infants are more vulnerable to hyperglycemia because they lack adequate insulin secretion from the pancreas and exhibit a measure of insulin-resistance [1,4]. Therefore, early recognition and diagnosis of neonatal NDM is rather challenging, particularly in premature infants.
NDM can be either transient or permanent. Transient NDM (TNDM) makes up about 50% of all NDM; this persists for a median of 12 weeks and resolves by 18 months. However, about 50% of patients relapse in late childhood or adolescence. Permanent NDM (PNDM) requires lifelong medical treatment; the disease does not resolve [1,4,5]. In terms of pathogenesis, relevant mutations are found in approximately 80% of neonates. Abnormalities of genes in the 6q24 locus and mutations of the ABCC8 or KCNJ11 genes encoding the potassium channel of the pancreatic beta-cell are known to cause NDM [1,2]. Oral sulfonylurea effectively treats both PNDM and TNDM [1,2,6].
Herein, we report a case of a premature infant with TNDM, the genetic cause of which was a variant of uncertain significance (VUS) in the ABCC8 gene; he was successfully treated with oral sulfonylurea.
CASE REPORT
A preterm male infant was delivered via cesarean section at 36 weeks of gestation to a 29-year-old mother. His Apgar scores were 9 at 1 minutes and 10 at 5 minutes. His birth weight, birth length, and head circumference were 2,120 g (3th–5th percentile), 44 cm (10th–25th percentile), and 31 cm (10th–25th percentile), respectively. As he exhibited intrauterine growth retardation, he was admitted to our neonatal intensive care unit for further evaluation. His father was Korean and his mother was Cambodian; there was no significant medical history of diabetes in his family. On the day of admission, his laboratory results were all in the normal ranges and no abnormalities were observed in physical examination.
On day of life (DOL) 3, full-bottle feeding was possible and his serum level of glucose was 106 mg/dL. He underwent routine blood testing before discharge on DOL 7; at that time, glucose levels were 272 mg/dL. He did not have any hyperglycemic symptoms, continued on full oral feeding, and exhibited good activity. We performed blood sugar tests 3 hours before several feedings, which revealed persistent hyperglycemia (from 150 to 287 mg/dL). On DOL 9, we considered the possibility of NDM and scheduled additional laboratory tests. The results were as follows: whole blood glucose, 248 mg/dL; sodium, 131 mmol/L; potassium, 5.4 mmol/L; chloride, 97 mmol/L; serum ketones, 0.1 mmol/L; insulin, 12.70 μIU/mL (reference range, 2–13 μIU/mL); C-peptide, 2.75 ng/mL (reference range, 1.06–3.53 ng/mL); and insulin-like growth factor I, 55.90 ng/mL (reference range, 12.6–90.4 ng/mL). He lacked metabolic acidosis and all diabetes-related autoantibody measurements and urine analyses were negative. Abdominal ultrasonography failed to find any structural abnormalities in the pancreas.
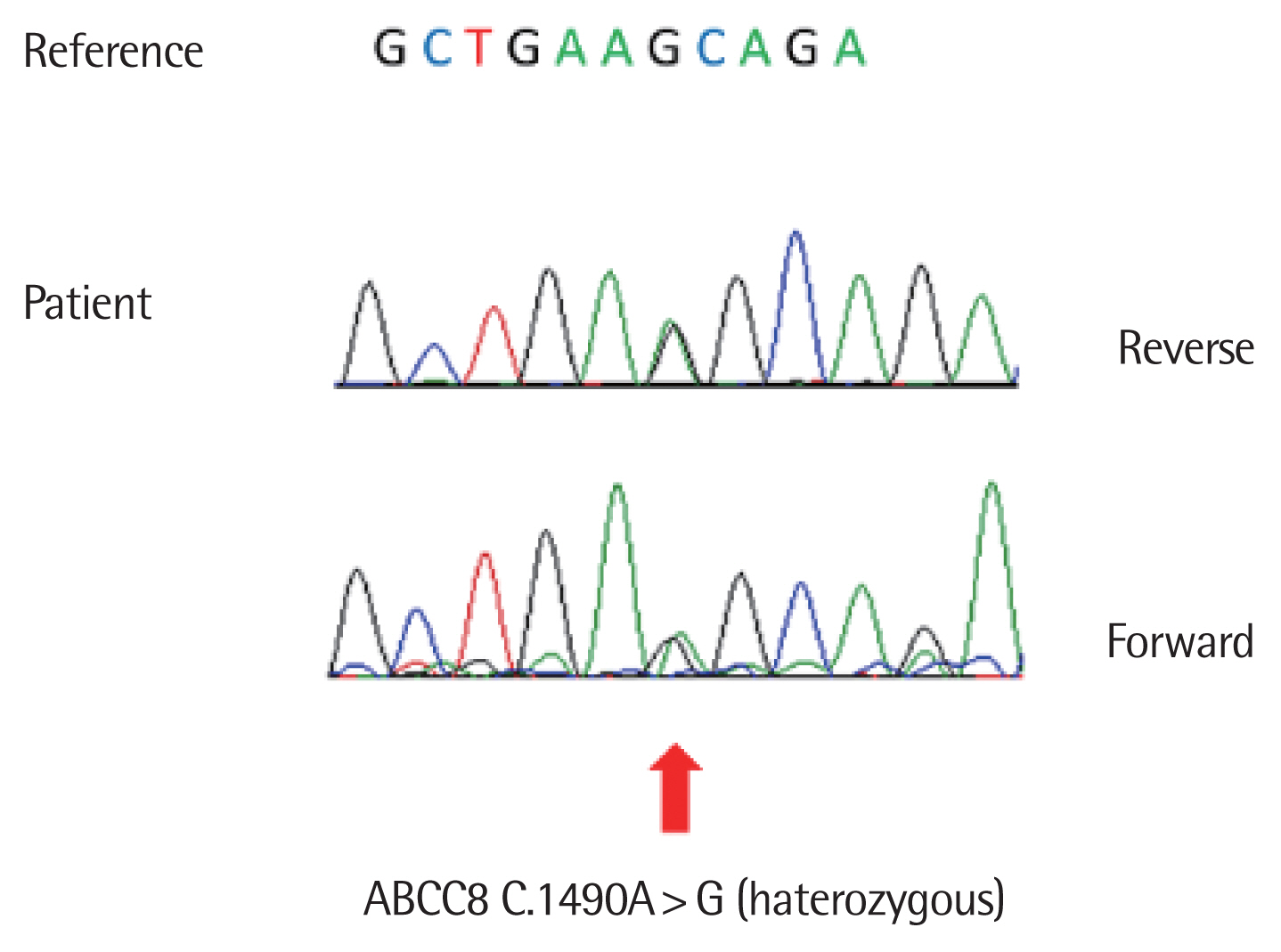
He was initially screened for abnormalities in chromosome 6q24; the results were negative. At the same time, we sought mutations in KCNJ11 and ABCC8. We found no pathogenic variant, but a VUS was evident in ABCC8 (c.1,490A>G mutation). This mutation is not in the current database (Fig. 1).
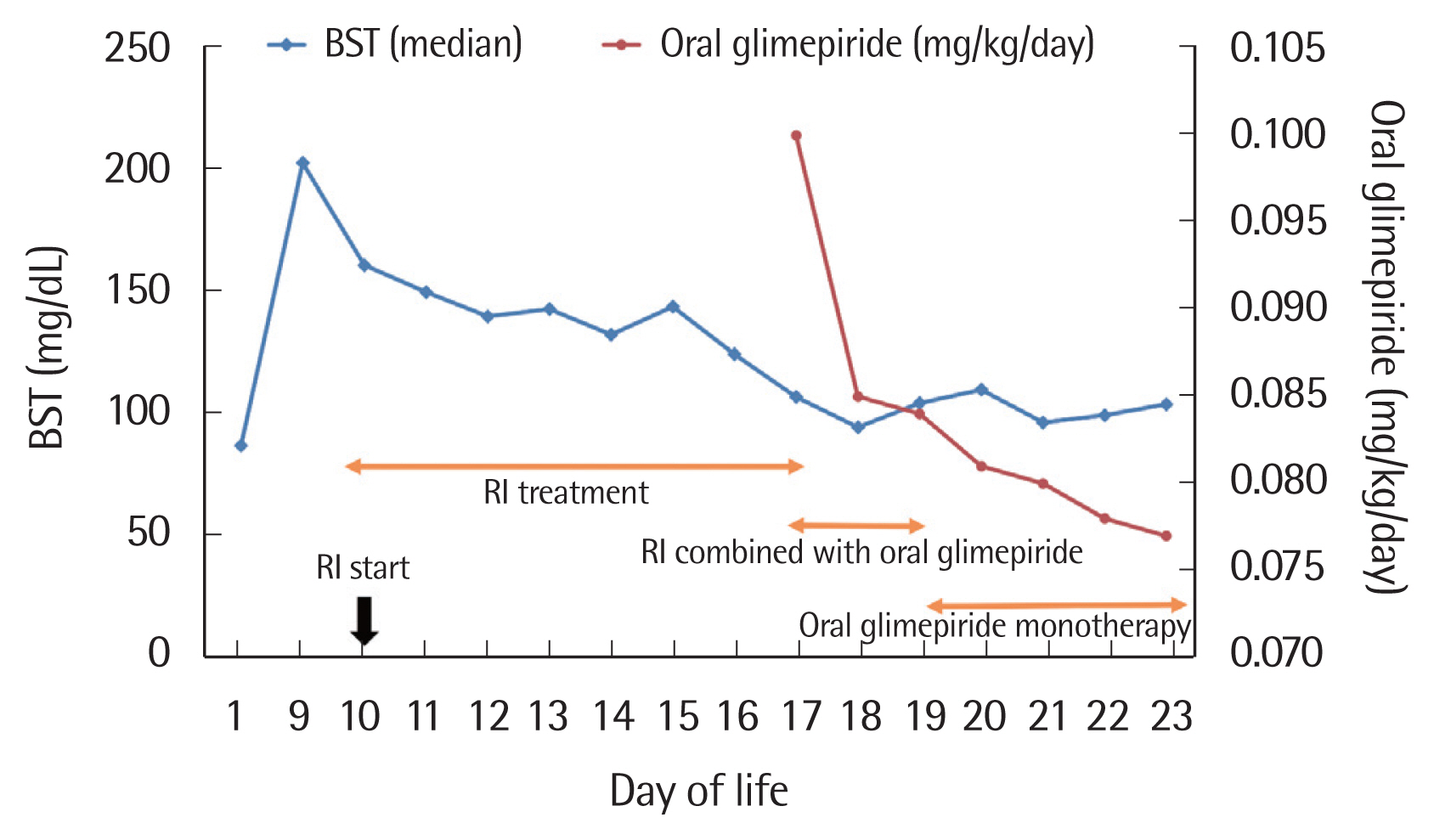
On DOL 10, we commenced subcutaneous injection of regular insulin (RI) (0.2 IU) twice daily 10 minutes prior to feeding. Blood levels of glucose were measured 4 hours before every feeding and 2 hours after each RI injection. Four days after commencement of insulin therapy, the afternoon insulin dose was reduced to 0.1 IU because the glucose level was well controlled. Both insulin doses were reduced to 0.1 IU after 6 days of insulin treatment. A transition to oral glimepiride (Amaryl; Handok Pharmaceutical Co., Seoul, Korea) 0.1 mg/kg once daily was attempted on DOL 17, 8 days after commencement of insulin therapy. The next day, RI was injected (0.1 IU) only once, and it was stopped on DOL 19. He was discharged on DOL 23 in healthy condition; the oral glimepiride (Amaryl) 0.1 mg/day was maintained until 2 months of life (Fig. 2). Since the oral glimepiride stopped, he did not have hyperglycemia. He did not experience hypoglycemic events during oral glimepiride treatment and is, as of writing, exhibiting normal growth and development. We have of course scheduled regular checkups.
This study adhered to the tenets of the Helsinki Declaration (http://www.wma.net/en/30publications/10policies/b3/) and was approved by the Research Ethics Committee of the Institutional Review Board (IRB) of Soonchunhyang University Cheonan Hospital (approval no., IRB 2021–04–016). Written informed consent was obtained from both parents.
DISCUSSION
Hyperglycemia in term neonates is defined as a blood glucose level above 150 mg/dL; insulin treatment is usually recommended when the blood sugar level is over 250 mg/dL [7]. NDM is diagnosed when persistent hyperglycemia begins within the first 6 months of life [1]. Neonatal sepsis, stress, medications such as steroids and catecholamines, parenteral glucose administration, prematurity, or low birth weight are well-known risk factors for neonatal hyperglycemia [1,7]. NDM symptoms vary, ranging from incidental asymptomatic hyperglycemia to severe dehydration. Small-for-gestational-age status caused by a lack of functional insulin, irritability, polyuria, and poor postnatal growth are common clinical manifestations of NDM [1].
NDM that develops prior to 6 months of age is usually attributable to an underlying monogenic defect; that which develops after 6 months may reflect an autoimmune disease [8]. To date, 22 genetic causes of NDM have been recognized but their clinical manifestations differ. Early recognition of NDM symptoms and genetic testing are necessary to predict the clinical prognosis and to facilitate familial genetic counseling if needed [2,9].
Although the clinical difference between transient and permanent neonatal diabetes does not always reflect distinct molecular mechanisms, overexpression of genes of the 6p24 locus is the most common cause of TNDM. Patients with 6p24 mutations are usually diagnosed earlier than those with KCNJ11/ABCC8 mutations; the former mutations are sometimes accompanied by macroglossia or an umbilical hernia [1,2]. The second most common cause of TNDM is a mutation in one of the genes encoding the two subunits of the adenosine triphosphate (ATP)-sensitive potassium channel (KATP-channel; encoded by KCNJ11 and ABCC8) of the pancreatic β-cell. This channel plays a key role in stimulating insulin secretion by pancreatic β-cells in response to the glucose level [1,2,4]. The common causes of PNDM are mutations in the KATP-channel- or INS-encoding genes [1,2,10]. Therefore, mutation of a KATP-channel gene can present clinically as either transient or permanent NDM. The ABCC8 gene, which encodes the sulfonylurea receptor 1 of the KATP-channel, causes about 10% of all NDM and about 66% of all TNDM (approximately 66%); most KCNJ11 mutations (approximately 90%) cause PNDM [10,11]. Several NDM cases caused by de novo ABCC8 gene mutations have been reported [11,12]. Our case was born small for gestational age, which is a common phenotype of NDM; he exhibited TNDM. Although he was premature, we did not hesitate to perform NDM genetic testing, as premature babies frequently exhibit monogenic diabetes. If an infant born after 32 full weeks of gestation evidences persistent hyperglycemia, genetic referral is recommended [13]. Direct sequencing of the ABCC8 gene revealed an A>G transition at nucleotide 1,490, which is not listed as a pathological variant in the current database. However, his TNDM symptoms could not be otherwise explained and oral sulfonylurea treatment was successful. Thus, we conclude that this variant is a de novo NDM mutation.
Sulfonylureas act on the KATP-channel to increase insulin secretion from pancreatic β-cells. In NDM patients with a mutation in the KCNJ11 or ABCC8 gene, insulin therapy can be replaced by oral sulfonylurea therapy, which effectively improves glycemic control [1,2,6]. We rapidly switched our patient from insulin therapy to oral glimepiride (Amaryl) because NDM is often associated with neuropsychological dysfunction and early, empirical, oral sulfonylurea therapy is associated with good neurodevelopmental outcomes [14,15]. We encountered no adverse effects of oral glimepiride, including transient diarrhea or tooth discoloration.
In summary, we report a premature infant with TNDM and a nonspecific ABCC8 mutation (1,490 A>G); he was successfully treated with oral sulfonylurea. Long-term close monitoring is planned (to puberty), with a focus on possible diabetes relapse.